
水热法制备高活性的纳米M oS2催化剂
2017-11-01 16:24李佳鹤王冬娥马怀军田志坚
石油化工 2017年9期
李佳鹤,王冬娥,马怀军,田志坚
(1.中国科学院 大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学,北京 100049)
水热法制备高活性的纳米M oS2催化剂
李佳鹤1,2,王冬娥1,马怀军1,田志坚1
(1.中国科学院 大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学,北京 100049)
采用水热法直接制备了Co/Ni促进的纳米MoS2催化剂,利用XRD,SEM,EDX-mapping和HRTEM方法对催化剂进行表征,并以二苯并噻吩(DBT)为模型化合物考察了加氢脱硫(HDS)性能。表征结果显示,该催化剂由纳米片层堆积而成,为典型的MoS2结构,纳米片尺寸约为5 nm,堆积度为1~2层,助剂元素均匀分布于MoS2中。催化剂直接用于催化DBT的HDS反应具有较高的活性,原料中n(Co)∶n(Co + Mo)= 0.3的催化剂活性最高,DBT转化率达到75.8%。催化剂在H2和H2/H2S氛围中热处理后,晶化度提高,活性位点数减少,HDS活性降低。添加Co助剂的催化剂的活性优于添加Ni助剂的催化剂。
加氢脱硫;非负载催化剂;助剂;二苯并噻吩;Co-Mo-S;水热法
Co/Ni促进的MoS2催化剂已广泛用于油品加氢和重油提质过程[1-2]。其中,负载型的催化剂一般用于馏分油加氢脱硫(HDS),采用固定床工艺[3-4]。而高分散的非负载催化剂则适用于悬浮床工艺,用于处理重质油组分[5-6]。随着原油的重质化以及环境法规的日益严格,开发悬浮床工艺以及高活性的非负载催化剂已成为研究的热点[7-8]。
含助剂的非负载MoS2催化剂的制备一般分为两步。首先,采用溶液反应、共沉淀法等制得过渡金属硫代盐或氧化态的催化剂前体,然后进一步将其在H2/H2S氛围中还原硫化,得到活化的Co/Ni促进的MoS2催化剂[9-11]。水热法操作简单,温度一般在200 ℃以下,条件较温和,水热反应直接制备的硫化态催化剂就可用于催化HDS反应,省略了在H2/H2S氛围中的高温硫化过程[12-13]。水热法制备非负载催化剂的合成参数、助剂种类以及所得催化剂的微观形貌对活性的影响还有待进一步研究。
本工作采用水热法直接制备了Co/Ni促进的纳米MoS2催化剂,利用XRD,SEM,EDX-mapping和HRTEM等方法对催化剂进行表征,并将其直接用于催化二苯并噻吩(DBT)的HDS反应,考察了合成参数及助剂种类对催化剂HDS活性的影响。
1 实验部分
1.1 催化剂的制备
水热法制备CoMo催化剂的步骤如下:按照n(Co)∶n(Co + Mo)= 0.1,0.3,0.5的计量比分别称取1.75 mmol四硫代钼酸铵(ATTM)和相应量的醋酸钴,与2 m L水合肼一起溶于30 m L去离子水中。将该溶液转移至100 m L反应釜中,200 ℃下水热反应24 h。产物过滤,用水和无水乙醇洗涤,70 ℃下真空干燥过夜,得到的试样记为CoMo-HT-x(x为原料中 n(Co)∶n(Co + Mo)的值)。
水热法制备NiMo催化剂的步骤同上,只是用相应量的醋酸镍代替醋酸钴,所得试样记为NiMo-HT-y(y为原料中 n(Ni)∶n(Ni+Mo)的值)。
催化剂的热处理:将水热法制备的CoMo-HT-x和NiMo-HT-y试样置于管式炉中,在H2气氛中400 ℃下保持2 h,所得试样分别标记为CoMo-H2-x和NiMo-H2-y。将水热法制备的CoMo-HT-x试样置于管式炉中,在10%(φ)H2S/H2混合气氛中400 ℃下保持2 h,所得试样记为CoMo-S-x。
1.2 催化剂的表征
XRD表征采用PANalytical公司Philips PANalytical X'Pert PRO型X射线衍射仪,Cu Kα靶(λ = 0.154 18 nm),管电压40 kV,管电流40 mA,扫描速率12.5(°)/min。SEM 和 EDX-mapping表 征 采 用JEOL公司JSM-7800F型高分辨扫描电子显微镜,加速电压20 kV;EDX-mapping测试Mo,S,Co元素的分布。HRTEM表征采用JEOL公司JEM-2100型透射电子显微镜,加速电压200 kV。
1.3 催化剂的活性评价
称量1.5 g的反应底物DBT、0.015 g催化剂、30 g正十二烷溶剂加入100 m L的Parr釜式反应器中。密闭釜体,用0.5~1.0 MPa的氢气置换釜中空气3次,随后填充氢气至8 MPa。调节搅拌转速为300 r/min,在350 ℃、自生压力下保持4 h。反应结束后卸压,取出反应物进行分析。
反应产物用微孔滤膜过滤掉其中的催化剂,采用装有HP-5型色谱柱的Agilent公司7890B-5977A型GC-MS气质联用装置定性分析其成分,采用装有HP-5型色谱柱和FID检测器的Agilent公司7890A型气相色谱仪进行定量分析。
2 结果与讨论
2.1 催化剂的表征结果
ATTM与醋酸钴或醋酸镍在水溶液中混合时,通过离子交换反应生成CoMoS4或NiMoS4,它们与还原剂水合肼在水热温度200 ℃下发生氧化还原反应,生成含Co或Ni的MoS2产物。催化剂的XRD谱图见图1。由图1可知,水热法制备的CoMo-HT催化剂均为弱晶化的MoS2。2θ为32°和58°左右的宽衍射峰可归属于2H-MoS2(JCPDS No.37-1492) 的(100) 和(110) 晶 面。 对 比MoS2的标准衍射图可发现,2θ = 14°左右的(002)晶面的衍射峰偏移至9°左右。MoS2为二维层状化合物,(002)晶面衍射峰的强度反应了层的堆积度,其位置表征了层间距的大小。根据布拉格方程2d sinθ = nλ,计算得到水热法制备的CoMo-HT催化剂层间距为0.98 nm,而正常的2H型MoS2的层间距为0.63 nm。这一现象在水热法制备的MoS2中较常见,文献一般认为是由于有小分子插入到MoS2的层间,导致层间距扩大所致,但关于插层物质的种类说法不一,主要有铵根离子、氧原子、水分子等[14-16]。本工作合成的催化剂体系中含铵根离子、水分子和氧离子,因而插层物质可能为以上任一种或几种的组合。CoMo-HT催化剂的所有衍射峰均存在宽化现象,且峰强度较弱,表明催化剂主要为弱晶化的MoS2相,粒径较小。XRD谱图中除了MoS2的3个宽衍射峰外,无其他衍射峰出现,表明催化剂中无单独的Co物种的晶相。
Yoosuk等[17-18]将采用相似的水热法制备的催化剂记为CoMoS2和NiMoS2,但该化学式并不能准确反应催化剂中各元素的计量比。催化剂中精确的原子计量比很难测定,为表征CoMo催化剂中Co元素分布情况,对CoMo-HT-0.3催化剂进行EDX-mapping表征,结果见图2。由图2可知,水热法制备的CoMo催化剂中,Co元素的分布与Mo和S元素相同,表明助剂元素均匀分布于MoS2中。
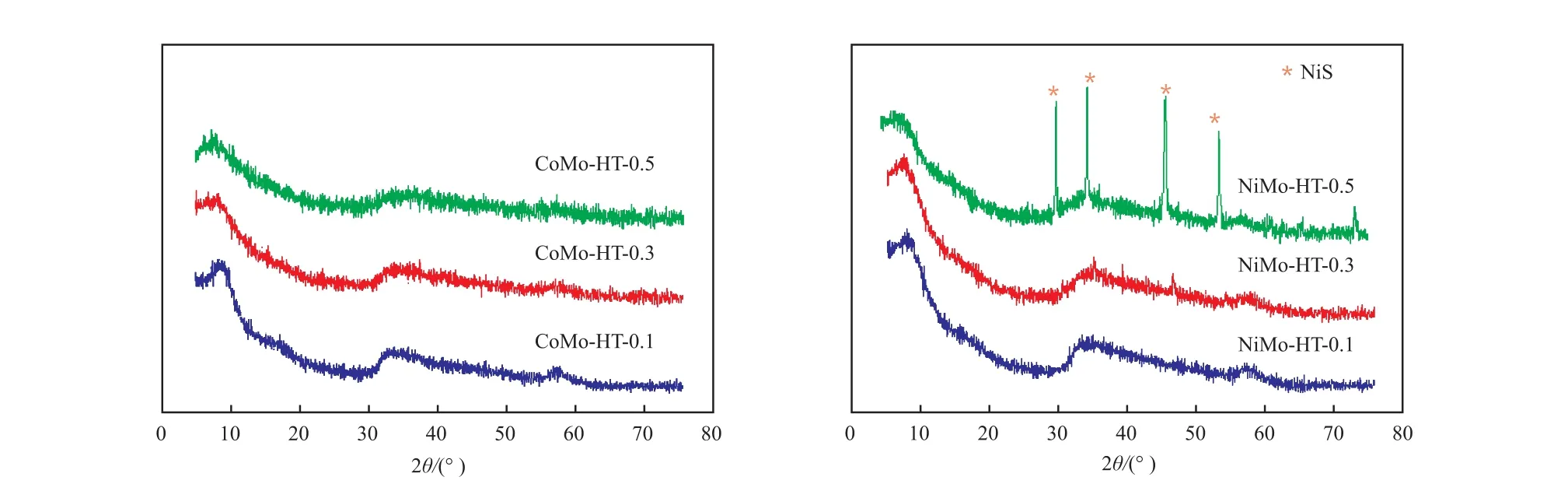
图1 催化剂的XRD谱图Fig.1 XRD patterns of the catalysts.
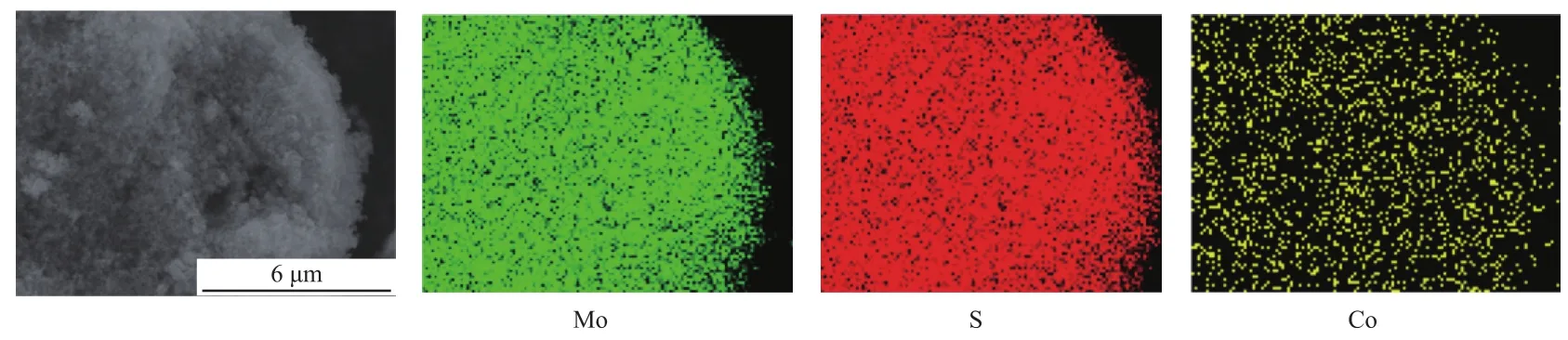
图2 CoMo-HT-0.3催化剂的EDX-mapping图Fig.2 EDX-mapping images of catalyst CoMo-HT-0.3.
水热法制备的NiMo-HT催化剂的XRD图谱与CoMo-HT的相似。助剂添加比n(Ni)∶n(Ni +Mo)为0.1和0.3时,谱图中只有归属于MoS2的3个宽衍射峰,表明催化剂为弱晶化的MoS2,无单独的Ni物种晶相。n(Ni)∶n(Ni + Mo)= 0.5时,谱图中除了归属于MoS2的宽衍射峰外,还出现了尖锐的衍射峰,这些尖衍射峰可归属为NiS相,表明在NiMo-HT-0.5催化剂中,部分Ni以NiS相存在。
H2和H2/H2S氛围中热处理后的催化剂的XRD谱图见图3。由图3可知,H2处理后的CoMo-H2催化剂在 2θ = 14°,32°,38°,58°的宽衍射峰可分别归属于2H型MoS2的(002),(100),(103)和(110)晶面,表明H2处理后不同助剂添加比的催化剂中均含有2H型MoS2。CoMo-H2-0.1和CoMo-H2-0.3的XRD谱图中只有MoS2的衍射峰,无Co物种的晶相;而CoMo-H2-0.5的XRD图中还出现了Co9S8的衍射峰,表明H2处理后CoMo-H2-0.5催化剂中部分Co元素转变为Co9S8相。与水热法制备的催化剂相比,H2处理后的催化剂的XRD谱图中归属于MoS2的宽衍射峰强度增强,表明MoS2晶化度增强。(002)晶面衍射峰移动到14°位置,这是由于高温处理过程去掉了H2O或铵根离子等插层小分子,导致层间距减小到0.63 nm。这种热处理导致的扩层现象消失在文献中也有报道,该扩层结构在后续的反应条件下很难重新生成[15]。在H2/H2S氛围中处理后的CoMo-S催化剂的XRD谱图与H2中处理的催化剂相似。CoMo-H2-0.1和CoMo-H2-0.3可归属于2H型MoS2,无Co物种晶相出现;CoMo-S-0.5中Co9S8的衍射峰强度更强,表明它在H2/H2S氛围中处理后生成的Co9S8晶化度更高。
水热法制备的NiMo催化剂在H2氛围处理后,归属于MoS2的宽峰强度增加,即MoS2晶化度提高。在NiMo-H2-0.1的XRD谱图中只有MoS2的衍射峰,无Ni物种晶相出现;在NiMo-H2-0.3和NiMo-H2-0.5的XRD谱图中除了MoS2的宽衍射峰外还出现了Ni3S2的衍射峰,表明H2处理后NiMo-H2-0.5催化剂中部分Ni元素转变为Ni3S2相。

图3 H2和H2/H2S中处理后的催化剂的XRD谱图Fig.3 XRD patterns of catalysts treated in H2 or H2/H2S.
CoMo-HT-0.3催化剂经不同气氛处理后的SEM照片见图4。由图4可知,CoMo-HT-0.3催化剂颗粒较小,尺寸在几十纳米;经H2和H2/H2S处理后,催化剂的微观形貌变化不大。

图4 CoMo-HT-0.3催化剂经不同气氛处理后的SEM照片Fig.4 SEM images of CoMo-HT-0.3 catalyst treated in different atmosphere.
CoMo-HT-0.3催化剂经不同气氛处理后的HRTEM照片见图5。由图5可知,CoMo-HT-0.3催化剂由纳米片层堆积而成,为典型的2H型MoS2结构,纳米片尺寸约为5 nm,堆积度为1~2层。在图5a中还观察到有层间距扩大的纳米片存在,这与XRD谱图预示的扩层结构相一致。在H2和H2/H2S中处理后,催化剂中纳米片的曲率减小,堆积度增加至3~5层,晶格条纹明显,表明催化剂的晶化度有较大提高。这与图3中衍射峰增强的结果相一致。

图5 CoMo-HT-0.3催化剂经不同气氛处理后的HRTEM照片Fig.5 HRTEM images of CoMo-HT-0.3 catalyst treated in different atmosphere.
2.2 HDS活性评价结果
以DBT为模型化合物考察了催化剂的HDS反应活性。图6为典型的DBT的HDS反应路径以及处理前后催化剂的活性图。从图6可看出,DBT的HDS有两条路径[19]:1)直接脱硫生成联苯(BP);2)先加氢生成四氢化二苯并噻吩(THDBT),再加氢脱硫生成苯基环己烷(CHB)。水热法制备的CoMo催化剂具有较高的HDS活性,DBT的转化率均在70%以上,这与催化剂的微观结构有关。MoS2是一种典型的二维层状化合物,在体相中,每个Mo原子与6个S原子键合,而在边缘位每个Mo原子仅与4个S原子结合,形成S空位。大量研究表明,MoS2边缘的S空位为催化反应的活性位[20-21]。Daage等[21]进一步研究了 HDS 反应中产物的选择性与MoS2催化剂中片层的堆积高度之间的关系,由此提出“rim-edge”模型:位于MoS2最上层和最下层的“rim”位点同时具有加氢和脱硫的作用,而位于中间层的“edge”位点则只有脱硫活性,无加氢活性。因而,少层数的MoS2由于具有更多的“rim”位点而具有较强的加氢与脱硫活性。与大尺寸、堆积层数多的块材MoS2相比,少层数、纳米级的MoS2可暴露更多的活性位,有利于催化活性的提高。本工作制得的CoMo催化剂其纳米片尺寸仅为5 nm左右,堆积度为1~2层,因而具有较高的催化活性。
水热法制备的CoMo催化剂进行HDS反应时,CoMo-HT-0.3催化剂的DBT转化率最高(75.8%),该结果与大多数负载型CoMo催化剂相同。Lauritsen[22-24]等曾通过穆斯堡尔谱和扩展X射线吸收精细结构谱等技术在CoMo催化剂中观测到一种活性相结构,并将其命名为“Co-Mo-S”相结构,该结构模型可较好地解释CoMo催化剂中Co的助剂效应。在该结构中,助剂Co原子位于MoS2的棱边,Co的量可以从无到覆盖整个MoS2的棱边。HDS活性依赖于Co-Mo-S相的数量,由此可知本工作中CoMo-HT-0.3催化剂中Co-Mo-S活性相的状态达到了最优。
由图6可知,CoMo-HT催化剂的转化率高于CoMo-H2和CoMo-S系列催化剂。这可能是由于水热法制备的CoMo-HT催化剂由弯曲的、不连续的纳米片组成,暴露出大量的缺陷位点。催化剂在H2和H2/H2S中热处理后,由无定形态转变为结晶度良好的状态,纳米片排列变得更有序,纳米片曲率变小,催化剂中缺陷位点数减少,从而导致HDS活性降低。此外,处理前后纳米片堆积层数的变化也会影响HDS活性。直接水热法制备的催化剂纳米片堆积层数为1~2层,处理后堆积层数增加至3~5层,导致催化剂中“rim”位点数减少,HDS活性有所降低。综上,采用水热法制备的CoMo-HT催化剂,避免了高温还原硫化处理过程,所得纳米片曲率较大、堆积层数少,保持了较高的HDS活性。

图6 DBT的HDS路径及CoMo催化剂的HDS活性Fig.6 Hydrodesulfurization(HDS) reaction path of dibenzothiophene(DBT) and HDS activities of CoMo catalysts.
含Co助剂的MoS2催化剂用于DBT的HDS反应时,主要产物为直接脱硫路径产生的BP与先加氢再HDS路径产生的THDBT和CHB。此外,BP,THDBT及CHB还可进一步发生加氢裂化或加氢异构反应生成甲基环戊烷(MCP)、苯和苄基环戊烷(BCP)。CoMo催化剂用于DBT的HDS反应时,选择性最高的产物均为直接脱硫路径产生的BP,表明含Co助剂的MoS2催化剂有助于直接脱硫路径的进行。直接脱硫路径每脱除一个S原子需消耗2个氢气分子,而先加氢再HDS路径则需消耗4个氢分子,因而采用CoMo催化剂可促进直接脱硫路径的进行,有效减少HDS反应的氢耗。CoMo-HT-0.3催化剂的BP的选择性最高,表明该催化剂的脱硫性能最好。CoMo-HT催化剂的裂化产物MCP和苯的选择性略高于H2和H2/H2S中处理后的催化剂,这可能是由于水热法制备的催化剂其纳米片层数更少,“rim”位点更多,因而加氢活性较高,容易进一步发生加氢裂化或异构反应。
考察了助剂种类对催化剂HDS活性的影响,结果见图7。由图7可知,在助剂添加比相同的情况下,CoMo催化剂的DBT转化率高于NiMo催化剂,表明Co助剂对HDS催化剂的助剂效应强于Ni助剂。对比选择性可知,采用CoMo-H2-0.3催化剂时,BP的选择性较高,表明DBT的直接脱硫占据主导作用,反应氢耗较低。采用NiMo-H2-0.3催化剂时,先加氢再HDS路径的产物THDBT和CHB的选择性大大增加,表明脱硫过程氢耗较大。对比可知,CoMo催化剂的HDS活性高于NiMo催化剂,且Co助剂有利于直接脱硫路径的发生而Ni助剂更有利于先加氢再HDS路径的进行。
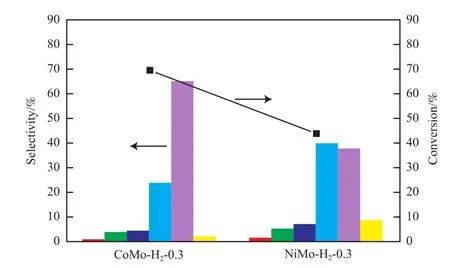
图7 助剂种类对催化剂HDS活性的影响Fig.7 Effects of different promoter on the HDS activities of catalysts.
3 结论
1)水热法直接制备的CoMo-HT催化剂由纳米片层堆积而成,为典型的MoS2结构,纳米片尺寸约为5 nm,堆积度为1~2层,助剂元素均匀分布于MoS2中。
2)水热法制备的CoMo催化剂直接用于催化DBT的HDS反应具有较高的活性。水热反应可直接得到硫化态的催化剂,可省略在H2/H2S氛围中高温还原硫化过程。CoMo-HT-0.3催化剂活性最高,DBT转化率达到75.8%。催化剂在H2和H2/H2S氛围中热处理后,晶化度提高,活性位点数减少,HDS活性降低。
3)添加Co助剂的催化剂的活性显著优于添加Ni助剂的催化剂,且Co助剂有利于直接脱硫路径的进行,反应氢耗较低;而Ni助剂则更有利于先加氢再HDS路径的发生,氢耗较大。
[1] Babich I V,M oulijn J A. Science and technology of novel processes for deep desulfurization of oil refinery streams:A review[J].Fuel,2003,82(6):607-631.
[2] Song Chunshan. An overview of new approaches to deep desulfurization for ultra-clean gasoline,diesel fuel and jet fuel[J].Catal Today,2003,86(1/4):211-263.
[3] 仝建波,蔺阳,刘淑玲,等. 加氢脱硫催化剂载体的研究进展[J].化工进展,2014,33(5):1170.
[4] 刘磊,吴新民. 加氢脱硫催化剂载体和活性组分的研究进展[J].工业催化,2008,16(8):1-7.
[5] Bellussi G,Rispoli G,Landoni A,et al. Hydroconversion of heavy residues in slurry reactors:Developments and perspectives[J].J Catal,2013,308:189-200.
[6] Zhang Shuyi,Liu Dong,Deng Wenan,et al. A review of slurry-phase hydrocracking heavy oil technology[J].Energy Fuels,2007,21(6):3057.
[7] Chianelli R R,Berhault G,Torres B. Unsupported transition metal sulfide catalysts:100 years of science and application[J].Catal Today,2009,147(3/4):275-286.
[8] 李贺,殷长龙,赵蕾艳,等. 非负载型加氢精制催化剂的研究进展[J].石油化工,2013,42(7):811.
[9] Olivas A,Zepeda T A,Villalpando I,et al. Performance of unsupported Ni(Co,Fe)/MoS2catalysts in hydrotreating reactions[J].Catal Commun,2008,9(6):1317-1328.
[10] Genuit D,A fanasiev P,Vrinat M. Solution syntheses of unsupported Co(Ni)-Mo-S hydrotreating catalysts[J].J Catal,2005,235(2):302-317.
[11] Olivas A,Galván D H,A lonso G,et al. Trimetallic NiMoW unsupported catalysts for HDS[J].Appl Catal,A,2009,352(1/2):10-16.
[12] 刘欢,殷长龙,赵蕾艳,等. 水热法合成Ni-M o非负载型催化剂及催化加氢性能[J].石油炼制与化工,2013,44(9):19-24.
[13] 钦柏豪,杨运泉,刘文英,等. 表面活性剂对水热法合成MoS2加氢脱硫催化剂的影响[J].燃料化学学报,2012,40(11):1384.
[14] Xie Junfeng,Zhang Jiajia,Li Shuang,et al. Controllable disorder engineering in oxygen-incorporated M oS2ultrathin nanosheets for efficient hydrogen evolution[J].J Am Chem Soc,2013,135(47):17881-17888.
[15] Li Jiahe,Wang Donge,Ma Huaijun,et al. Ionic liquid assisted hydrothermal synthesis of hollow core/shell MoS2m icrospheres[J].Mater Lett,2015,160:550-554.
[16] Zhang Chaofeng,Wang Xu,Li M ingrun,et al. Chemoselective transfer hydrogenation to nitroarenes mediated by oxygenimplanted MoS2[J].Chin J Catal,2016,37(9):1569.
[17] Yoosuk B,Kim J H,Song Chunshan,et al. Highly active MoS2,CoMoS2and NiMoS2unsupported catalysts prepared by hydrothermal synthesis for hydrodesulfurization of 4,6-dimethyldibenzothiophene[J].Catal Today,2008,130(1):14-23.
[18] Yoosuk B,Song Chunshan,Kim J H,et al. Eff ects of preparation conditions in hydrothermal synthesis of highly active unsupported NiMo sulfide catalysts for simultaneous hydrodesulfurization of dibenzothiophene and 4,6-dimethyldibenzothiophene[J].Catal Today,2010,149(1/2):52-61.
[19] 刘丽,郭蓉,孙进,等. 柴油加氢脱硫催化剂的研究进展[J].化工进展,2016,35(11):3503.
[20] Lauritsen J V,Nyberg M,Nørskov J K,et al. Hydrodesulfurization reaction pathways on MoS2nanoclusters revealed by scanning tunneling microscopy[J].J Catal,2004,224 (1):94-106.
[21] Daage M,Chianelli R R. Structure-function relations in molybdenum sulfide catalysts:The “Rim-Edge” model[J].J Catal,1994,149(2):414-427.
[22] Lauritsen J V,Helveg S,Lægsgaard E,et al. Atom ic-scale structure of Co-Mo-S nanoclusters in hydrotreating catalysts[J].J Catal,2001,197:(1):1-5.
[23] Lauritsen J V,Bollinger M V,Lægsgaard E,et al. Atomicscale insight into structure and morphology changes of MoS2nanoclusters in hydrotreating catalysts[J].J Catal,2004,221(2),510-522.
[24] Lauritsen J V,Kibsgaard J,Olesen G,et al. Location and coordination of promoter atoms in Co- and Ni-promoted MoS2-based hydrotreating catalysts[J]. J Catal,2007,249(2):220-233.
Hydrothermal synthesis of M oS2nano-catalysts with high activities
Li Jiahe1,2,Wang Donge1,Ma Huaijun1,Tian Zhijian1
(1. Dalian Institute of Chem ical Physics,Chinese Academy of Sciences,Dalian Liaoning 116023,China;2. University of Chinese Academy of Sciences,Beijing 100049,China)
Co/Ni promoted M oS2nano-catalysts were synthesized by hydrothermal method directly and characterized by XRD,SEM,EDX-mapping and HRTEM. hydrodesulfurization(HDS)performances of catalysts were evaluated by using dibenzothiophene(DBT) as model compound. The results showed that the catalysts were built by nanosheets,which were typical structure of MoS2.The slab lengths of nanosheets were about 5 nm and stacking layer numbers were 1-2. The promoted elements distributed in MoS2evenly. As-synthesized catalysts had high activity when used to catalyze HDS reaction of DBT directly. When the atom ic ratio of Co to (Co+Mo) was 0.3 in the raw materials,the conversion of dibenzothiophene achieved the highest to 75.8%. After heat treatment under H2or H2/H2S atmospheres,the crystallization degree of catalysts was enhanced and the amount of active sites decreased,which resulted in the decrease of HDS activity. Co-promoted catalysts had higher activity than Ni-promoted catalysts.
hydrodesulfurization;unsupported catalysts;promoter;dibenzothiophene;Co-Mo-S;hydrothermal
1000-8144(2017)09-1125-07
TQ 426.95
A
2017-03-15;[修改稿日期]2017-06-14。
李佳鹤(1990—),女,河南省许昌市人,博士生,电话 0411-84379768,电邮 lijiahe@dicp.ac.cn。联系人:田志坚,电话0411-84379151,电邮 tianz@dicp.ac.cn。
中科院战略性先导专项基金项目(XDA07020300);国家自然科学基金项目(21303186)。
10.3969/j.issn.1000-8144.2017.09.005
(编辑 邓晓音)
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
今日农业(2020年20期)2020-11-26
石油石化绿色低碳(2019年6期)2019-02-13
潍坊学院学报(2016年2期)2016-12-01
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
当代化工研究(2016年7期)2016-03-20
中国资源综合利用(2016年4期)2016-01-22
橡胶工业(2015年8期)2015-07-29
中国洗涤用品工业(2015年5期)2015-02-28
