
全外显子及靶向文库捕获测序在多囊肾病基因诊断中的应用比较
2017-09-20 03:16刘维强张慧敏李浩贤孙筱放
分子诊断与治疗杂志 2017年5期
刘维强 张慧敏 李浩贤 孙筱放
•论 著•
全外显子及靶向文库捕获测序在多囊肾病基因诊断中的应用比较
刘维强 张慧敏 李浩贤 孙筱放★
目的比较全外显子和靶向文库捕获高通量测序对多囊肾病相关基因的检测效率。方法对6份多囊肾病标本(包括一份低比例嵌合变异标本)分别进行全外显子捕获或靶目标捕获2种方法建立文库,Illumina HiSeq 2000仪器连续双向测序。结果以PKD1、PKD2和PKHD13个基因作为目标序列,靶向捕获法平均测序深度为190倍,5倍以上测序深度占全部有效序列的85.59%,靶区域覆盖度95%以上,但PKD1第一外显子仍有200~300 bp区域不能覆盖;全外显子捕获法平均测序深度为28.34倍,5倍以上测序深度占全部有效序列的55.35%,目标区域覆盖度较低,PKD1外显子覆盖度小于40%。结论靶向文库捕获测序法具有较髙的敏感性、准确性,更适合PKD基因变异的检测,但高通量测序技术对PKD1基因检测仍有不足之处。
多囊肾病;靶向捕获;全外显子
作者单位:广州医科大学附属第三医院,广东省产科重大疾病重点实验室,广东省普通高校生殖与遗传重点实验室,广东,广州510150
多囊肾病(polycystic kidney disease,PKD)是指在肾脏中发生多个充满液体的囊肿并导致肾脏结构和功能损害的疾病,这种囊肿还可以累及到其他器官[1]。根据遗传方式不同,其又分为常染色体显性遗传多囊肾病(autosomomal domainant polycystic kidney disease,ADPKD)和常染色隐性遗传多囊肾病(autosomomal recessive polycystic kidney disease,ARPKD)[2]。PKD 疾病严重影响着病人的生活质量和生命健康。
PKD主要致病基因为PKD1、PKD2和PKHD1基因。PKD1、PKHD1是分别有46个和67个外显子的大基因,另外PKD1基因存在假基因,且PKD疾病相关基因突变不存在热点,因此,对其分子诊断非常困难并且检测非常费时、费力且技术要求高。目前基于全外显子和目标序列捕获的下一代高通量测序技术(next generation sequencing,NGS)已开始应用于PKD等遗传性疾病的分子诊断[3⁃5]。本研究对 PKD患者标本应用全外显子和目标序列文库捕获2种方法测序,比较这2种方法在PKD疾病诊断的效率。
1 材料与方法
1.1 对象
选择PKD1、PKD2、PKHD13个基因为靶目标区域进行效率评估。选取6份样本进行全外显子捕获或靶目标文库捕获方法进行测序。为更好地评估这2种方法对PKD疾病的检测效率,本研究选取的样本中包括一例变异位点为低比例嵌合的ADPKD患者标本。
1.2 试剂和仪器
DNA提取方法采用DNeasy Tissue试剂盒(Qiagen公司,德国),DNA打断仪Covaris S2系统购自美国Covaris公司,全外显子捕获和靶目标捕获2种文库建立方法分别选用美国Agilent公司的SureSelect试剂和美国Roche公司的定制基因片段捕获芯片,测序试剂和仪器来自美国Illumina公司。
1.3 文库构建及测序
3 μg基因组DNA经Covaris S2系统打断为200~250 bp大小片段,分别使用美国Agilent公司的SureSelect测序试剂进行全外显子文库构建和美国Roche公司定制的基因片段捕获芯片进行靶目标文库富集。文库经Agilent 2100 Bioanalyzer和ABI StepOne进行片段大小、浓度、富集度的检测,最后利用高通量测序仪Illumina HiSeq 2500 Analyzers(Illumina,美国)连续双向测序 90个循环,用 Illumina Pipeline software(version 1.3.4)读出原始测序数据。
1.4 数据分析
数据下机后进入信息分析部分。首先对下机的原始数据(raw reads)进行测序质量评估,去除低质量以及被接头污染的reads。随后用BWA软件(Burrows Wheeler Aligner)与GRCh37/Hg19进行序列比对,与此同时进行序列捕获效果评价,用SOAP⁃snp软件和Samtools软件分别进行SNV(single nu⁃cletide variant)和 Indel(insertion and deletion)的查询,生成目标区域碱基多态性结果,随后进行数据库的比对,应用NextGENe软件找出可疑突变。
2 结果
2.1 文库质量评价
文库质量经Tap⁃Stations检测,文库片段大小、分子量均符合要求。
2.2 全外显子捕获法和靶目标捕获法NGS有效数据分析
通过NextGENe软件对原始数据的处理,全外显子捕获法测序平均得到50 482 664条有效序列读数,其中与基因组参考序列比对上的序列平均有49 938 146条序列,占全部有效序列的98.92%。以PKD1(NM_001009944)、PKD2(NM_000297)、PKHD1(NM_138694)3个转录本外显子作为目标靶序列比对,平均得到14 122条有效靶序列读数,占全部有效序列的0.03%。平均测序深度为28.34倍,5倍以上测序深度占全部有效序列的55.35%。
靶目标捕获法NGS测序平均得到47 928 884条有效序列读数,其中与基因组参考序列比对上的序列平均有47 173 064条序列,占全部有效序列的98.42%。用以上3个基因转录本外显子作为目标靶序列比对,平均得到173 559条有效靶序列读数,占全部有效序列的0.23%。平均测序深度为190倍,5倍以上测序深度占全部有效序列的85.59%,结果如图1。
2.3 全外显子捕获与靶目标捕获法对PKD1基因测序的数据比较
通过比较2种不同方法对PKD1基因的捕获效率,本研究发现靶目标捕获法在覆盖度和测序深度上均要比全外显子捕获法好,见图2。
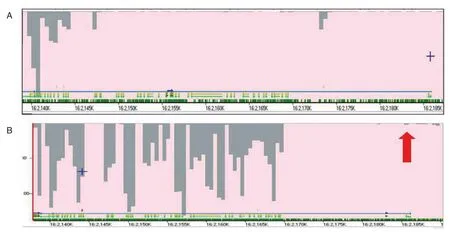
图2 2种不同方法对PKD1基因的捕获效率Figure 2 Evaluation the capture efficiency ofPKD1gene between the 2 methods
2.4 全外显子捕获与靶目标捕获法对PKD2基因测序的数据比较
通过比较2种不同方法对PKD2基因的捕获效率,发现在覆盖度和测序深度上靶目标捕获法与全外显子捕获法效率没有明显差异,见图3。
2.5 全外显子捕获与靶目标捕获法对PKHD1基因测序的数据比较
通过比较2种不同方法对PKHD1基因的捕获效率,发现在覆盖度和测序深度上靶目标捕获法与全外显子捕获法效率没有明显差异,见图4。
2.6 对测序未覆盖区域的分析
虽然靶目标捕获法相对全外显子捕获法有较好的测序深度和测序覆盖度,但仍有部分区域如PKD1第1外显子不能完全覆盖。本研究对这部分未覆盖区域进行了分析,以明确缺失区域具体位置,方便设计引物进行常规Sanger测序弥补。利用NextGENe软件可以清楚地发现未覆盖区域的具体位置,见图5。
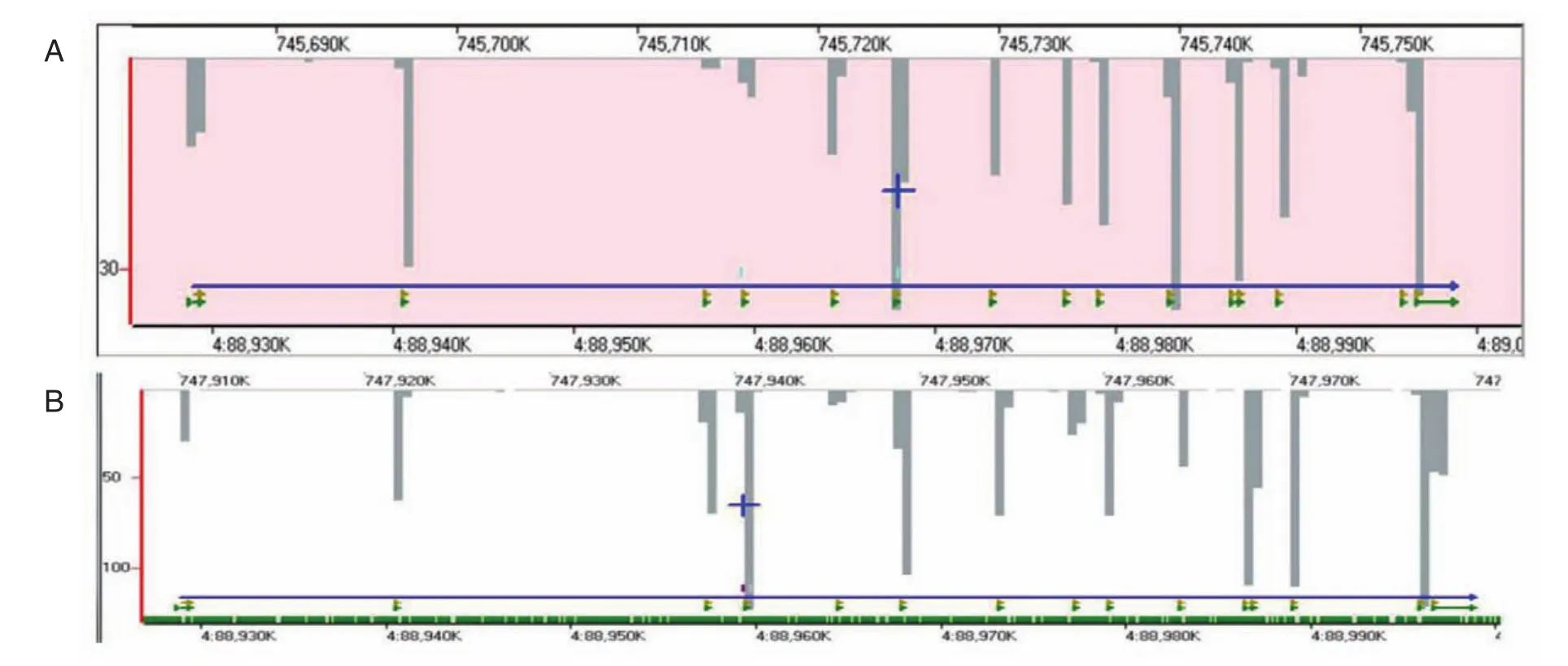
图3 2种不同方法对PKD2基因的捕获效率Figure 3 Evaluation of the capture efficiency ofPKD2gene between the 2 methods
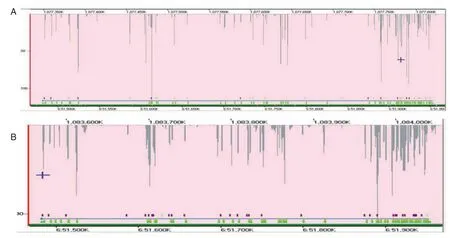
图4 2种不同方法对PKHD1基因的捕获效率Figure 4 Evaluation of the capture efficiency ofPKHD1 gene between the 2 methods
2.7 低比例嵌合变异NGS结果分析
本研究所选一例样本在16号染色体2168790号位置发生了错义突变(c.416G>A,p.Trp139Ter),存在低比例嵌合变异现象。利用全外显子捕获测序法,由于此区域整个测序深度只有5倍左右,导致嵌合突变没有有效检出。利用靶目标捕获测序(targeted capture sequencing),此区间测序浓度达到1 830倍,虽然软件自动分析时将此嵌合突变过滤掉,但由于测序深度足够大,经调整参数设置,此错误过滤得以纠正,成功检测出此低比例嵌合,如图6。
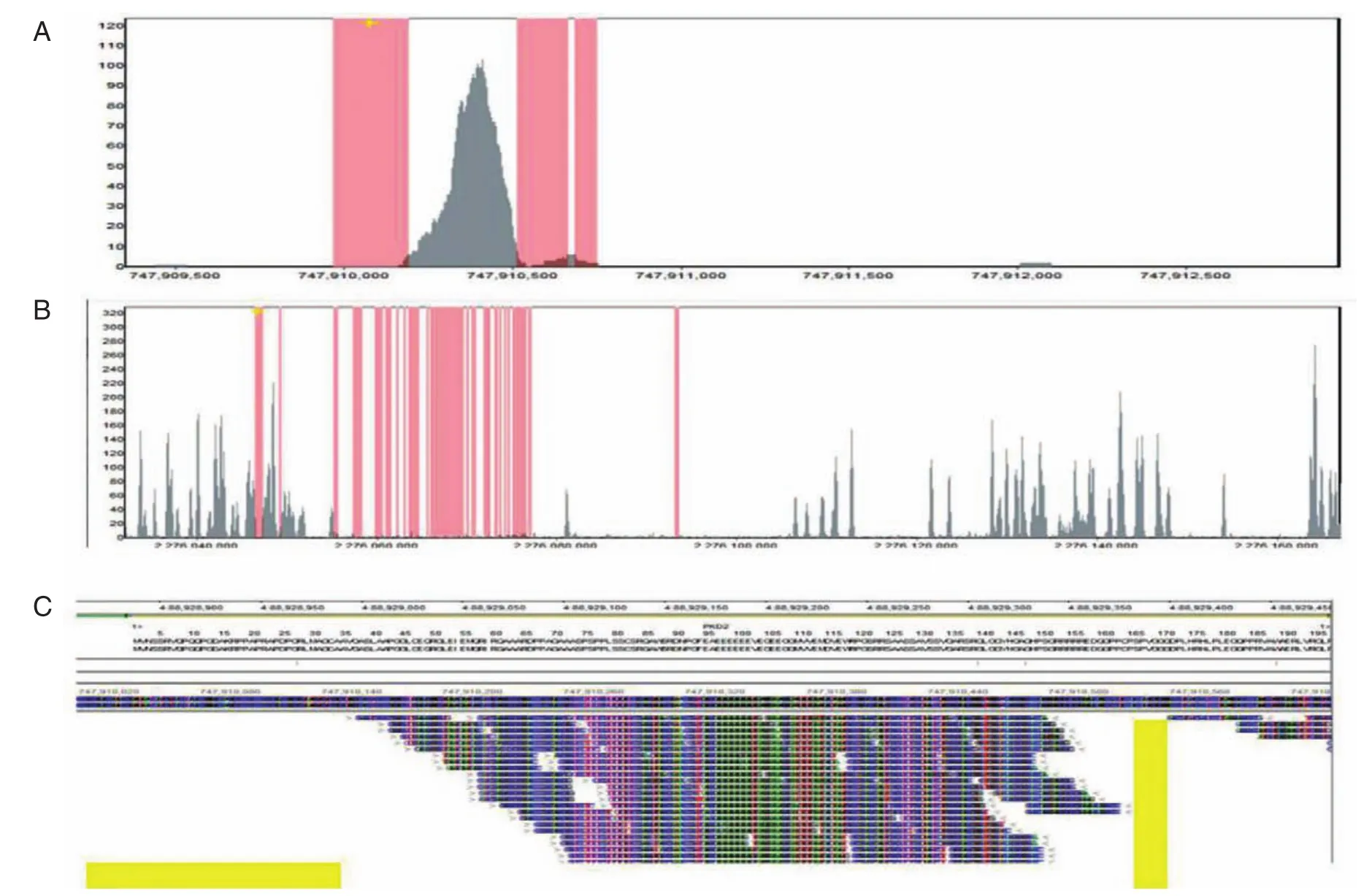
图5 NGS测序数据未覆盖区域的定位Figure 5 Identify the uncovered data regions after NGS
3 讨论
PKD疾病基因检测目前常用传统的Sanger测序法,其最主要的缺点就是测序通量太低,一次PCR实验只能检测1个到2个外显子,而PKD疾病由3个基因组成,需测序的外显子总数加起来有120多个,使得整个诊断体系效率偏低。另外,Sanger测序还存在着扩增失败、DNA交叉污染等因素,所有这些因素均导致整个流程成本高、时间长、工作量巨大[6],使得这种基于传统技术基础上的诊断体系很难满足目前临床检测的需求。
NGS技术具有高通量、测序速度快、准确性高、结果稳定等特点。目前利用NGS技术全外显子组测序、目标序列靶向测序的科研和临床应用已广泛开展[3,7⁃8]。基于不同的文库构建方法,NGS应用较为普遍的3种模式分别为全基因组测序(whole genome sequencing,WGS)[7]、全外显子测序(whole exome sequencing,WES)[8]及靶目标捕获测序[9]等。目前,NGS技术已应用于PKD疾病的分子诊断[10⁃12]。
最近有文献比较了全外显子和靶目标测序方法对肿瘤样本的检测效率,发现靶目标测序方法相对全外显子测序方法在结果准确度上更加可靠[13]。针对多囊肾病,本研究比较了这2种NGS检测方法的检测效率。虽然全外显子捕获方法相对靶目标测序方法成本偏高,但其可以在全基因范围进行外显子的检测,可以发现除PKD1、PKD2、PKHD1外潜在的新的与多囊肾相关的基因或与肾病相关的基因变异[14]。本研究发现虽然全外显子捕获方法在PKD1基因的检测效率低于靶目标捕获方法,但其在PKD2和PKHD1基因的捕获效率上与靶目标捕获方法没有太大差异。因此当患者具有典型的多囊肾表型而常规3个基因又没有发现变异的情况下可以考虑使用此方法。
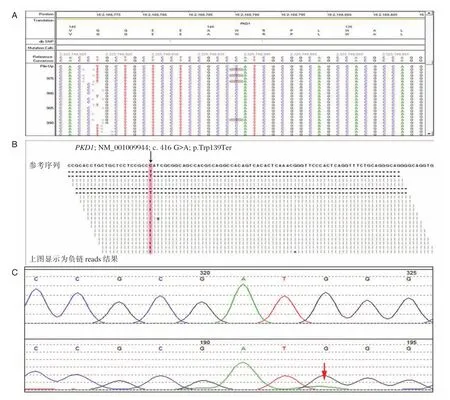
图6 NGS检测嵌合变异Figure 6 Detection the mosaic mutation by using NGS
相对于全外显子捕获测序,虽然靶目标捕获测序捕获的外显子数目较全外显子捕获大幅减少,但其对目标区域的测序深度有效提高,因此这种方法有足够量的测序读数进行变异的分析。在本研究中,我们应用Roche公司定制的基因捕获芯片进行靶目标的捕获。这款定制芯片可以靶向捕获222个基因的3 093个外显子以及剪切位点,目标基因涵盖多囊肾病的3个致病基因。通过Next⁃GENe软件对原始数据的预处理,本研究证实靶目标捕获法测序得到有效序列读数、5倍以上测序深度占全部有效序列比例等多个关键指标都要比全外显子捕获效率更高。分析原因,笔者认为,全外显子捕获PKD1基因效率低的主要原因是由于全外显子捕获探针对部分基因尤其是GC含量特别高的基因如PKD1的捕获敏感性很差,探针不能有效与目标区域结合,直接导致目标区域没有数据。同时由于基因组中存在大量假基因[15],很大一部分此区域数据被认为是测序错误或样本污染而被软件自动过滤掉了,导致最终可用的有效数据大大减少。
靶目标捕获测序方法测序深度的增加也有利于发现低比例嵌合变异。以本研究一例低比例嵌合变异为例,虽然嵌合比例只有15%左右,但由于靶目标捕获在此区域测序深度高达1 830倍,因此仍能灵敏地被检测到。即便如此,NGS方法仍有其不足之处,如对高GC含量的区域NGS方法的捕获效率明显偏低[16],这与本研究发现PKD1第一外显子有数百碱基序列没有覆盖到,第42号外显子的测序深度相对其他区域要低很多现象一致。分析原因可能与文库构建时高GC含量导致PCR扩增效率低有关[17]。对于此部分数据,本实验利用分析软件明确了未覆盖或低覆盖区域的确切区域及片段大小,可利用一代测序进行补全。
总之,通过比较全外显子捕获方法和靶目标捕获法测序在多囊肾基因变异诊断中的应用,本研究发现靶目标捕获法对于多囊肾基因诊断具有更高的性价比和更高的捕获效率。当然,NGS技术对复杂结构基因、高GC含量区域的检测仍有不足之处,需结合常规Sanger测序进行相互补充。
[1]Harris PC,Torres VE.Genetic mechanisms and signal⁃ing pathways in autosomal dominant polycystic kidney disease[J].J Clin Invest,2014,124(6):2315⁃2324.
[2]Ghata J,Cowley BD,Jr.Polycystic kidney disease[J].Compr Physiol,2017,7(3):945⁃975.
[3]Edrees BM,Athar M,Al⁃Allaf FA,et al.Next⁃genera⁃tion sequencing for molecular diagnosis of autosomal recessive polycystic kidney disease[J].Gene,2016,591(1):214⁃226.
[4]Liang WC,Tian X,Yuo CY,et al.Comprehensive tar⁃get capture/next⁃generation sequencing as a second⁃tier diagnostic approach for congenital muscular dystrophy in Taiwan[J].PLoS One,2017,12(2):e0170517.
[5]Yang T,Meng Y,Wei X,et al.Identification of novel mutations of PKD1 gene in Chinese patients with auto⁃somal dominant polycystic kidney disease by targeted next⁃generation sequencing[J].Clin Chim Acta,2014,433:12⁃19.
[6]Liu W,Chen M,Wei J,et al.Modification of PCR conditions and design of exon⁃specific primers for the efficient molecular diagnosis of PKD1 mutations[J].Kidney Blood Press Res,2014,39(6):536⁃545.
[7]Chen R,Feng Y,Wang X,et al.Whole genome se⁃quences of three Clade 3 Clostridium difficile strains carrying binary toxin genes in China[J].Sci Rep,2017,7:43555.
[8]Zhou T,Souzeau E,Sharma S,et al.Whole exome se⁃quencing implicates eye development,the unfolded pro⁃tein response and plasma membrane homeostasis in pri⁃mary open⁃angle glaucoma[J].PLoS One,2017,12(3):e0172427.
[9]Feng J,Li Y,Jia Y,et al.Spectrum of somatic muta⁃tions detected by targeted next⁃generation sequencing and their prognostic significance in adult patients with acute lymphoblastic leukemia[J].J Hematol Oncol,2017,10(1):61.
[10]Kinoshita M,Higashihara E,Kawano H,et al.Techni⁃cal evaluation:identification of pathogenic mutations in PKD1 and PKD2 in patients with autosomal domi⁃nant polycystic kidney disease by next⁃generation se⁃quencing and use of a comprehensive new classification system[J].PLoS One,2016,11(11):e0166288.
[11]Mallawaarachchi AC,Hort Y,Cowley MJ,et al.Whole⁃genome sequencing overcomes pseudogene ho⁃mology to diagnose autosomal dominant polycystic kid⁃ney disease[J].Eur J Hum Genet,2016,24(11):1584⁃1590.
[12]Borras DM,Vossen R,Liem M,et al.Detecting PKD1 variants in polycystic kidney disease patients by single⁃molecule long⁃read sequencing[J].Hum Mutat,2017,38(7):870⁃879.
[13]Chang YS,Huang HD,Yeh KT,et al.Evaluation of whole exome sequencing by targeted gene sequencing and sanger sequencing[J].Clin Chim Acta,2017,471:222⁃232.
[14]Braun DA,Schueler M,Halbritter J,et al.Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with child⁃hood ⁃onset increased renal echogenicity[J].Kidney Int,2016,89(2):468⁃475.
[15]Zhang Z,Gerstein M.Large⁃scale analysis of pseudo⁃genes in the human genome[J].Curr Opin Genet Dev,2004,14(4):328⁃335.
[16]LaDuca H,Farwell KD,Vuong H,et al.Exome se⁃quencing covers>98%of mutations identified on target⁃ed next generation sequencing panels[J].PLoS One,2017,12(2):e0170843.
[17]Choi M,Scholl UI,Ji W,et al.Genetic diagnosis by whole exome capture and massively parallel DNA se⁃quencing[J].Proc Natl Acad Sci USA,2009,106(45):19096⁃19101.
Comparison of the application of whole exome capture sequencing and targeted capture sequencing methods in gene diagnosis of polycystic kidney disease
LIU Weiqiang,ZHANG Huimin,LI Haoxian,SUN Xiaofang★
(Key Laboratory for Reproduction and Genetics of Guangdong Higher Education Institutes,Key Laboratory for Major Obstetric Diseases of Guangdong Province,Third Affiliated Hospital of Guangzhou Medical University,Guangzhou,Guangdong,China,510150)
ObjectiveTo evaluate the efficiency of the whole exome capture sequencing method and the targeted libraries capture sequencing method for improving the efficiency of molecular diagnosis of polycystic kidney disease.MethodsThe libraries of 6 tested samples(including 1 sample with low ratio mosaic mutation)were prepared by whole exome capture method and targeted capture method,respectively.Illumina HiSeq2000 was used for sequencing.ResultsThe selected target sequence werePKD1,PKD2andPKHD1.The average depth in targeted capture method and whole exome sequencing method is 190 and 28.34,respectively.The ratio of region of target sequence with 5x coverage is 85.59%and 55.35%in targeted capture method and whole exome sequencing method,respectively.Coverage analysis indicated that the average coverage of whole exome sequencing method is less than 40%,which is significantly lower than that of targeted capture method(95%);however,200~300 base pair of the first exon ofPKD1uncovered yet using targeted capture method.ConclusionTargeted capture method is more suitable forPKDgene⁃based diagnostic using high⁃throughput sequencing.However,the uncovered region still needs conventional Sanger sequencing for complement.
Polycystic kidney disease;Targeted capture;Whole exome
广东省科技厅项目(2014A020212354,2013B051000087,201400000004⁃4,201400000003⁃4);广东省医学科研基金(A2015327)
★通讯作者:孙筱放,E⁃mail:xiaofangsun@gzhmu.edu.cn
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
猪业科学(2021年3期)2021-05-21
江苏卫生保健(2021年9期)2021-03-27
家庭医药(2021年2期)2021-03-09
中国生殖健康(2020年4期)2021-01-18
保健医苑(2020年6期)2020-12-04
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
中国生殖健康(2018年4期)2018-11-06
猪业科学(2018年4期)2018-05-19
