
Leber先天性黑曚患者临床特征与基因筛查
2017-09-19 02:20董嫱,张华
转化医学电子杂志 2017年8期
董 嫱,张 华
(1济南市第八人民医院,山东济南250013;2中国医学科学院,北京协和医学院,北京100730)
Leber先天性黑曚患者临床特征与基因筛查
董 嫱1,张 华2
(1济南市第八人民医院,山东济南250013;2中国医学科学院,北京协和医学院,北京100730)
目的:对Leber先天性黑矇的临床表现以及遗传学特点予以分析,并对已知的和可能存在的致病基因突变位点进行筛查研究.方法:以56例Leber先天性黑矇患者为研究对象,收集其临床资料.使用多聚酶链式反应技术(PCR)将Lerber先天性黑矇致病相关致病基因视网膜色素上皮基因(RPE65)和卵磷脂视黄醇酰基转移酶基因(LRAT)中的全部外显子与外显子-内含子接会处进行扩增,测序分析致病的突变基因.结果:对56例患者行RPE65基因十四个外显子和LRAT基因三个外显子的检测,发现了7处单核苷酸多态性改变.结论:由于Leber先天性黑曚的临床表现具有多样性,其诊断有赖于症状和眼底表现以及各项辅助检查.本组56例患者可能与RPE65外显子和LRAT外显子突变无关.不同患者均发现同一位点的同样单核苷酸多态性的改变,具有一定的临床价值和基础研究意义,应增加样本量对蛋白功能进一步分析.
视网膜色素上皮基因;卵磷脂视黄醇酰基转移酶基因;Leber先天性黑曚;突变;基因
0 引言
Leber先天性黑矇(leber congenital amurasis,LCA)属于一种少见的常染色体隐性遗传病,也有少数的显性遗传报道.其可导致严重的视网膜病变,约占遗传性视网膜变性病变的5%,并且发病早,可导致10%~20%的儿童失明[1-2].LCA的临床表现呈多样性,可导致婴幼儿先天性失明,即出生时或出生后不久出现严重的视觉丧失,有畏光、眼球震颤、固视不能、指压眼球等临床表现,眼底早期可无明显异常,随病程进展眼底周围可出现椒盐样的色素沉积,视网膜血管狭窄,引起脉络膜和视网膜色素上皮细胞的萎缩.视网膜电图(electroretinogram,ERG)检测到波幅平缓甚至消失等表现.除此之外,患者常伴有神经系统功能异常、肥胖、代谢系统疾病等严重的并发症,还可能出现圆锥角膜、白内障等异常表现[3-4].目前研究显示,LCA可由包括编码视网膜色素上皮特异性65 kD 蛋白[retinal pigment epithelial(cell)65,RPE65]、磷脂酰胆碱在卵磷脂-视黄醇酰基转移酶(lecithin retinol acyltransferase,LRAT)[5]在内的多种不同基因突变所致.LCA具有遗传的异质性,所编码的蛋白质是催化全反式视黄醇转变成11-顺式视黄醇的异构酶之一,后者再经过一系列代谢转变成视紫红质,参与光信号的传导过程.11-顺视黄醛在视网膜色素上皮层(retinal pigment epithelium,RPE)细胞内经过一系列酶促反应由全反式视黄醇转变而来.通过内吞作用摄取感光细胞内的全反式视黄醇和从脉络膜中吸收的类胡萝卜素经过转化而变为全反式视黄醇两种方式,再转变成为RPE细胞内的全反式视黄醇[6].类胡萝卜素从脉络膜转运至 RPE后,在RPE65的催化下转变为全反式视黄醛,后者在与LRAT的催化下经过酯化反应生成全反式视黄酯,进而在视黄酯水解酶(retinyl ester hydrolase,REH)与视黄醇结合蛋白 1(cellular retinol-binding protein,CRBP1)的催化下生成全反式视黄醇.RPE65基因和LRAT基因都是导致LCA的常见基因.本文应用PCR对RPE65以及LRAT基因进行突变基因的筛查分析,对LCA的病因产生机制进行更为深入的了解,为临床中该病的诊断提供帮助.
1 资料和方法
1.1 一般资料 本研究以位于北京、泰安、兰州盲校的LCA患者以及在北京协和医院眼科门诊治疗的散在患者共计56例为研究对象.所有研究对象经检查排除其他的遗传性疾病或先天性异常.
1.2 方法 对56例研究对象采集外周静脉血进行DNA提取,筛查突变基因.本实验所涉及到的RPE65和LRAT两个基因的引物由上海生工生物技术公司合成,其分子量计算公式如下:MW=(A碱基数× 312)+(C碱基数×288)+(G碱基数×328)+(T碱基数×303)-61.将RPE65与LRAT基因的引物序列送至上海生工生物技术有限公司进行合成.引物合成后,应用PCR技术对两个基因的外显子及外显子-内含子交接区域进行DNA扩增,对PCR产物使用2%的聚丙烯酰胺琼脂糖凝胶电泳进行鉴定分析.然后将上述扩增产物转运至北京华大基因研究中心以及天一辉远生物科技有限公司,以PCR产物进行扩增DNA,提纯DNA后再进行基因测序.
应用 NCBI的 BLAST 2 seqences和 UCSC的BLAT search,对比Genbank中的原始基因序列和测序结果的序列,分析已获取的序列图谱,从而确认是否存在基因突变.如果检测到存在基因突变,则首先对该位点进行单核苷酸多态性分析,确认该位点是否为已知的正常多态位点;然后查阅国内外相关研究文献,确认该位点是否已经过研究报道;最后,若确认为新的突变位点,则仍需对至少50名与该患者无任何血缘关系的正常人进行该位点的PCR检测、测序,以确认是否为未知群体多态性所致.
2 结果
2.1 临床表现 收集北京协和医院眼科门诊治疗的15例LCA患者的临床资料,包括患者初诊时的年龄、性别及符合诊断标准的主要临床症状(畏光、夜盲)、视力、屈光状态、眼球内陷、指眼征、眼底检查、初始诊断(表1).

表1 15例LCA患者的临床资料
2.2 Leber先天黑曚RPE65基因的突变结果 所有研究对象的测序结果中均未发现基因异常序列,但发现其中存在4处单核苷酸多态性的改变(表2).

表2 RPE65基因检测的单核苷酸多态性改变
测序结果显示56例患者中有一例患者的RPE65基因中的3号外显子的cDNA278位的甘氨酸被谷氨酸所取代(G278A),密码子由GGA变为GAA.RPE65基因exon-3 PCR产物测序结果(部分)见图1.
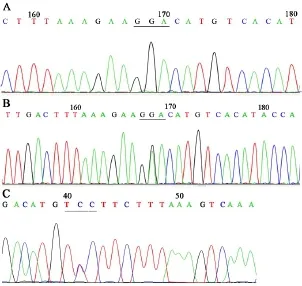
图1 RPE65基因3号外显子的测序结果
在RPE65基因中10号内含子的改变为GAA 352 GAG,56名受检者中共发现了26名有该改变.RPE65基因exon-10 PCR产物测序结果(部分)见图2.
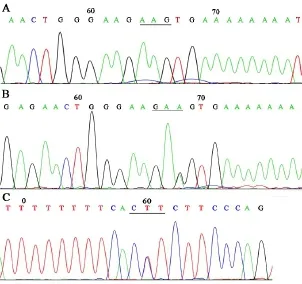
图2 RPE65基因10号外显子的测序结果
在RPE65基因中12号内含子的改变为IVS12+ 20a/c,56名受检者中共发现了 27名有该改变.RPE65基因exon-12 PCR产物测序结果(部分)见图3.

图3 RPE65基因12号外显子的测序结果
在RPE65基因中14号内含子的改变为C^A Ins AG,56名受检者中共发现了9名有该改变.RPE65基因exon-14 PCR产物测序结果(患者正向测序结果)见图4.

图4 RPE65基因14号外显子的测序结果
2.3 Leber先天黑曚LRAT基因的突变结果 所有研究对象的测序结果中均未发现基因异常序列,但发现其中存在2处单核苷酸多态性的改变(表3).

表3 LRAT基因检测的单核苷酸多态性改变
56例患者中有5例测序结果显示在LRAT基因中2a号内含子的改变为Ivs2a-12t/c.LRAT基因exon-2a PCR产物测序结果(部分)见图5.
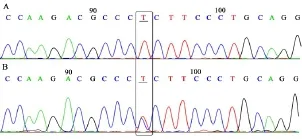
图5 LRAT基因2a号外显子的测序结果
56例患者中发现11例测序结果显示在LRAT基因中2b号内含子的改变为Ivs2b-55c/a.LRAT基因exon-2b PCR产物测序结果(部分)见图6.
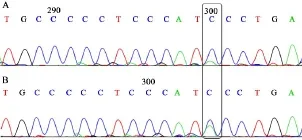
图6 LRAT基因2a号外显子的测序结果
3 讨论
LCA是一种少见的视网膜遗传性疾病,通常发病早、病情重,具有临床表现的多样性和遗传异质性,病因与多种因素有关.根据目前的研究显示,14个已知的基因和尚未明确与发现的多种基因的突变均与该病的发生有关.其中RPE65与LRAT基因都是引起LCA的常见突变基因.RPE65基因编码RPE65蛋白,该蛋白是一种视网膜色素上皮细胞特有的特异性蛋白,分子量为65 kDa,位于LCA2位点(lp31),广泛表达于视网膜色素上皮细胞中.该蛋白主要参与维生素A的代谢以及视紫红质的再生.因此RPE65突变的LCA患者易早期出现视锥细胞光感受器的缺失,说明该基因同时参与视觉生色团的产生,对视锥细胞的正常发育有重要作用.而当早期出现明显的视锥细胞光感受器缺失后,残余结构仍能维持多年的视锥结构和功能,由此可以推断,存在着其他重要的旁路可以影响视锥细胞光感受器的生存.LRAT是由230多个氨基酸组成的多肽,分子量为26 kDa,其具有视黄醇催化合成功能,且能使细胞从循环中获得视黄醛并存储于肝星状细胞的脂滴以及RPE的视网膜样结构中.LRAT位于内质网膜,研究认为其具有一个单跨膜的拓扑结构,N-端位于胞浆内,C-端位于内质网腔内.真核细胞中,C-端的跨膜区对于LRAT的活性与内质网的靶向功能都具有十分重要的作用,而N-端的疏水区则对于内质网膜的靶向功能或者酶活性的作用通常是非必须的,并且其氨基酸序列属于非保守序列[7-8].
LCA动物模型的建立,有利于更好地理解基因缺陷在视网膜细胞死亡中的作用,并且找到治疗的方法.近年来关于RPE65的基因治疗在动物实验上的应用取得显著成效,为LCA的临床治疗提供了新的研究方向[9-10].RPE65基因治疗仅在第一期临床试验中取得良好的成效,可能是因为该突变主要位于视网膜色素上皮细胞,易于药物的转入[11].当前,LCA (RPE65基因突变所致)基因治疗项目已经获得美国政府及其他非营利性基金会的援助.在北美以及欧洲,与RPE65基因突变相关的LCA患者已经能够获得基因治疗,其余基因突变型也处于临床试验阶段,为广大患者提供了康复愿景.并且我国也加入到国际合作项目LCA基因治疗的3期临床研究中,可以为前期确诊的患者提供免费的基因治疗.
目前在国内对于LCA的误诊率很高,对该病进行更多的认识和了解具有重要的临床意义.LCA给患者、其家庭及整个社会带来的负担是巨大的.LCA致病基因的发现为认识视网膜的发育、生物化学、细胞学机制以及研究视网膜变性发病的机制提供了新的信息.成功的开展RPE65基因突变所导致的LCA基因治疗的临床试验,具有划时代的意义,给患者带来了希望.本文通过对56例患者进行临床分析和基因测序后发现多处同一位点的同样单核苷酸多态性的改变.我们仍需收集更多的LCA病例,进一步研究中国LCA患者的基因型和临床表型的相关性,为将来开展基因治疗提供依据.
[1]Scholl HP,Moore AT,Koenekoop RK,et al.Safety and proof-ofconcept study of oral QLT091001 in retinitis pigmentosa due to inherited deficienciesofretinalpigmentepithelial 65 protein (RPE65)or lecithin:retinol acyltransferase(LRAT)[J].PLoS One,2015,10(12):e0143846.
[2]Cideciyan AV.Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy[J].Prog Retin Eye Res,2010,29(5):398-427.
[3]Mackay DS,Borman AD,Sui R,et al.Screening of a large cohort of leber congenital amaurosis and retinitis pigmentosa patients identifies novel LCA5 mutations and new genotype-phenotype correlations[J].Hum Mutat,2013,34(11):1537-1546.
[4]睢瑞芳,赵 潺,董方田,等.Leber先天黑矇的临床研究[J].中华眼底病杂志,2009,25(6):443-446.
[5]Leber T.Ueber retinitis pigmentosa und angeborene Amaurosis[J].Archiv für Ophthalmologie,1869,15(3):1-25.
[6]Koenekoop RK,T raboulsi EI.Leber's congenital amaurosis,stargardt disease,and pattern dystroph ies.In:T raboulsi EI.Genetic D iseases of the Eye[M].New York:Oxford University Press Inc 1998:3732387.
[7]Rozet JM,Perrault I,Gerber S,et al.Complete abolition of the retinal-specific guanylyl cyclase(retGC-1)catalytic ability consistently leads to leber congenital amaurosis(LCA)[J].Invest Ophthalmol Vis Sci,2001,42(6):1190-1192.
[8]Ikeda A,Naggert JK,Nishina PM.Genetic modification of retinal degeneration in tubby mice[J].Exp Eye Res,2002,74(4):455-461.
[9]Zernant J,Külm M,Dharmaraj S,et al.Genotyping microarray (disease chip)for Leber congenital amaurosis:detection of modifier alleles[J].Invest Ophthalmol Vis Sci,2005,46(9):3052-3059.
[10]den Hollander AI,Roepman R,Koenekoop RK,et al.Leber congenital amaurosis:genes,proteins and disease mechanisms[J].Prog Retin Eye Res,2008,27(4):391-419.
[11]Perrault I,Delphin N,Hanein S,et al.Spectrum of NPHP6/ CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype[J].Hum Mutat,2007,28(4):416.
Clinical characteristics and genetic screening of patients with Leber congenital amaurosis
DONG Qiang1,ZHANG Hua2
1The Eighth People's Hospital of Jinan,Jinan 250013,China;2Chinese Academy of Medical Sciences,Peking Union Medical College,Beijing 100730,China
AIM:To analyze the clinical manifestations and genetic characteristics of Leber's congenital amaurosis(LCA)and investigate the known and potential pathogenic gene mutation sites.METHODS:A total of 56 cases of LCA patients were selected as the objects of study,and their clinical data were analyzed.The retinal pigment epithelium gene 65(RPE65)and the leucovide retinol acyltransferase gene(LRAT)are the members of LCA.All exons and connections between exon and intron of these two genes RPE65 and LRAT were amplified by polymerase chain reaction(PCR),then their sequences were detected and analyzed to explore the related mutant genes.RESULTS: Fourteen exons of RPE65 gene and three exons of LRAT gene were detected in 56 patients,and seven single nucleotide polymorphisms were found.CONCLUSION:Because of the diversified clinical manifestations of LCA,the diagnosis depends on the symptoms,fundus manifestations and the auxiliary examinations.A total of 56 patients with LCA in the study may be irrelevant to exons'mutation of RPE65 and LRAT.However,we found that the same changing site of the single nucleotide polymorphisms were contained in all the 56 patients.This finding provide potential clinical values for the disease of LCA.
retinal pigment epithelium gene;lecithin retinol acyltransferase gene;Leber congenital amaurosis; mutation;gene
Q344+.12
A
2095-6894(2017)08-20-04
2017-01-19;接受日期:2017-02-03
科技部国际合作项目(2010DFB33430);美国防盲协会(CD-CL-0808-0470-PUMCH)
董 嫱.E-mail:155189814@qq.com
张 华.博士,副研究员.研究方向:眼科学.E-mail:zhanghua.eye@163.com
猜你喜欢
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
中学生数理化(高中版.高考理化)(2020年1期)2020-11-24
山地农业生物学报(2020年2期)2020-11-09
现代农业科技(2020年15期)2020-08-16
江苏农业科学(2019年6期)2019-09-25
浙江农业学报(2019年6期)2019-06-24
中国畜牧杂志(2019年5期)2019-01-12
医药前沿(2019年18期)2019-01-04
中国社区医师(2017年27期)2018-02-24
华人时刊(2017年21期)2018-01-31
