
1例疑诊Leigh综合征患儿及其父母线粒体相关基因突变观察
2017-08-09 01:27:44李亚丽孙艳美张宁王方娜高健
山东医药 2017年27期
李亚丽,孙艳美,张宁,王方娜,高健
(河北省人民医院,石家庄050071)
1例疑诊Leigh综合征患儿及其父母线粒体相关基因突变观察
李亚丽,孙艳美,张宁,王方娜,高健
(河北省人民医院,石家庄050071)
目的 观察并分析1例疑诊Leigh综合征患儿及其父母的线粒体相关基因突变情况。方法 发育落后合并肌张力异常、疑诊Leigh综合征的患儿1例,3岁,患儿就诊时其母孕14周。采集患儿及其父母的外周血2 mL,提取血液基因组DNA,采用高通量测序分析检测患儿线粒体病相关基因,并观察其父母基因突变情况。根据检测出的患儿线粒体相关基因突变位点,在患儿母亲孕18周时抽取羊水,采用PCR法检测胎儿是否存在相同突变位点,分析胎儿患病的风险。结果 测得患儿过量位点蛋白1(SURF1)基因有两个突变位点c.655G>T(p.E219X)和c.324-1G>C,c.655G>T来源于母亲、c.324-1G>C来源于父亲,为复合杂合突变,确诊患儿为疑诊Leigh综合征。患儿母亲羊水PCR检测分析发现胎儿为c.324-1G>C突变,无c.655G>T突变,为杂合突变携带者,胎儿出生后随访至1岁,孩子身体健康,能平稳独自行走。结论 发现Leigh综合征患儿SURF1基因的两个突变位点c.655G>T(p.E219X)、c.324-1G>C,是患儿发育落后、肌张力异常的致病基因;患儿父母均为杂合基因突变携带者,遗传方式是常染色体隐性遗传,母亲每次受孕其胎儿患病概率为25%。
线粒体肌病;Leigh综合征;过量位点蛋白1基因;基因突变;常染色体隐性遗传
线粒体是真核细胞中的一种细胞器,它是细胞内氧化磷酸化和合成三磷酸腺苷(ATP)的主要场所,为细胞95%的活动提供所需能量,并且它还参与细胞分化、细胞信息传递和细胞凋亡等过程,有调控细胞生长和细胞周期的能力。线粒体有自身的遗传物质和遗传体系,线粒体基因约编码人类2%的蛋白质[1]。线粒体肌病是由于线粒体结构和功能异常造成细胞呼吸链及能量代谢障碍的临床综合征,当其伴有中枢神经系统症状时称为线粒体脑肌病。线粒体肌病较常见的一种临床类型是Leigh综合征(LS),多发生于婴幼儿或儿童期,起病较隐匿,呈慢性进行性的肌张力倒退、伴随运动和智力发育迟缓,部分患者可出现呼吸衰竭、运动障碍而危及生命。早期诊断和治疗LS非常重要。该病临床表现多样,单纯依靠临床表现和一般实验室检查很难准确诊断,目前诊断该病的金标准是基因诊断[2~4]。本研究回顾分析了对1例临床疑似LS患儿的分子遗传学研究过程,发现了过量位点蛋白1(SURF1)基因的两个新的突变位点,明确了诊断和病因,并对其母孕期进行了产前诊断,现报告如下。
1 资料与方法
1.1 临床资料 疑似LS患儿1例,女,3岁,发育落后。生后9个月始能独坐,1岁3个月可以扶走,扶走时双下肢抬起困难,至今不能独自站立和行走;仅能简单发音“爸爸、妈妈”;1岁3个月运动倒退,四肢无力,呕吐、喂养困难,易哭闹、睡眠差,血乳酸3.0 mm/L;头MRI检查示双侧基底节区及脑干异常信号,部分病变弥散受限。根据患儿临床症状和检验结果疑诊LS。患儿父母同龄(25岁),身体均健康,否认家族中有遗传病史。患儿母亲25岁,23岁足月顺产此患儿,为第1次孕产,患儿生后Apgar评分正常。患儿母亲现孕14周,为避免再次生育此类患儿,要求行产前诊断。
1.2 线粒体相关基因突变观察 采集患儿及其父母的外周血2 mL(EDTA抗凝),按照血液基因组DNA提取试剂盒说明书提取基因组DNA(天根试剂盒)。以电泳和NanoDrop超微量分光光度计检测DNA的质量和浓度。提取1 500 ng的基因组DNA,超声打断成平均300 bp的长度,进行文库构建;连接8 bp的index标签和接头到打断的DNA上;连接好接头的片段化DNA与内分泌疾病捕获芯片进行杂交捕获;捕获后的文库质量和浓度通过片段分析和定量PCR进行评估。上illumina HiSeq X ten平台测序,基因组中佝偻病相关基因的测序数据量在2 G左右,平均测序深度在100 X。测序数据通过NextGENe软件进行数据过滤和比对分析。筛选的突变点在数据库(dbSNP、ExAC和1000 genome projects)中比对来确定人群携带率,进一步判断其致病性;通过SIFT、Polyphen-2 and mutation taster等软件进行突变点保守性及致病性分析,预测其致病性。对疑似致病的突变位点,通过一代测序进行验证,测序结果借助Mutation Surveyor v4.0软件与参考序列进行比对。
1.3 患儿母亲羊水细胞中线粒体相关基因突变观察 患儿母亲于孕18周时抽取羊水,采用PCR法检测胎儿是否存在相同突变位点,进行产前诊断。于产后1年对胎儿出生后的情况进行随访。
2 结果
测得患儿SURF1基因有两个突变位点c.655G>T(p.E219X)和c.324-1G>C,c.655G>T来源于母亲、c.324-1G>C来源于父亲,为复合杂合突变(见图1、2),确诊患儿为LS。患儿母亲羊水PCR检测分析发现胎儿存在c.324-1G>C突变,未发现c.655G>T突变,为杂合突变携带者(见图1、2)。患儿母亲足月顺产一男婴,现已1岁,身体健康,能平稳独自行走。
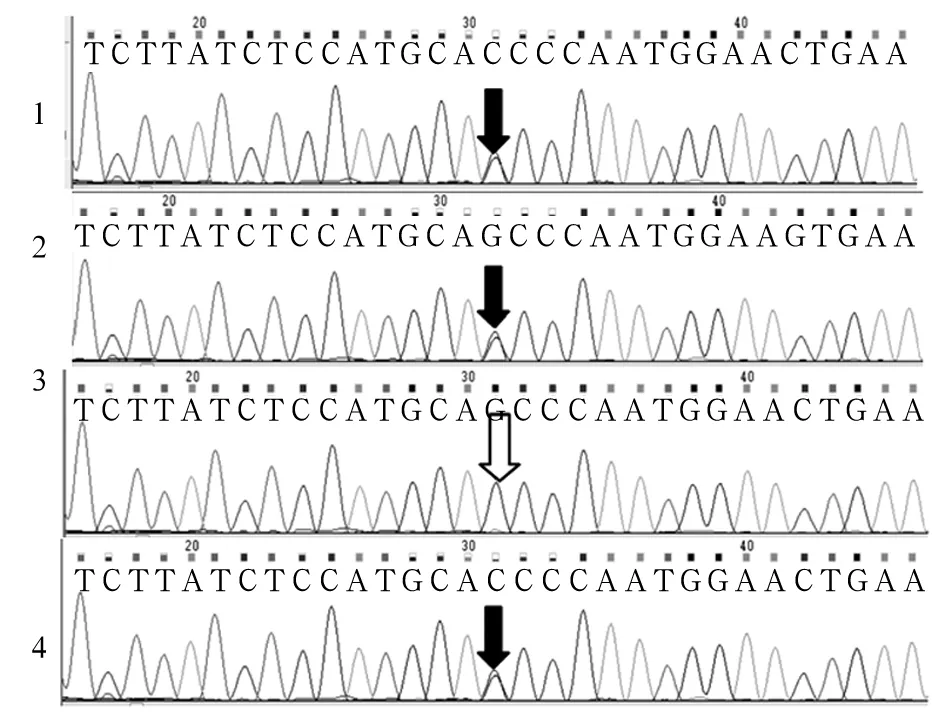
注:1为患儿样本;2为患儿父亲样本;3为患儿母亲样本;4为羊水细胞样本;黑色箭头示SURF1基因突变位点c.655G>T(p.E219X);白色箭头处SURF1基因不存在突变位点c.655G>T(p.E219X)。
图1 SURF1基因突变位点c.655G>T(p.E219X)
3 讨论
LS最早由英国学者[5]发现并描述其神经病理学特征。LS是多种酶缺陷导致的线粒体呼吸链氧化磷酸化障碍,引起退行性中枢神经系统损害,其特征是脑干、基底节和脊髓后柱区有对称性局灶坏死,故又称为亚急性坏死性脑脊髓病[6,7]。LS病因复杂,临床发病率约1/40 000。LS按患者发病年龄可分为三种类型,即新生儿型、经典婴儿型、青少年型。其中以婴儿型最常见,临床表现不一,包括运动和智力低下(包括舞蹈症)、肌张力减退、痉挛、小脑共济失调等,发病后2年内病死率高[8~10]。LS患者血清或脑脊液中乳酸含量增高[11~12]。祝小芬等[13]研究认为静息时乳酸升高、运动后即刻超过正常值3倍可作为拟诊LS的参考。LS头部MRI检查特征性表现是基底节尤其壳核区可见多灶性、双侧对称性的脑软化灶。病理学研究发现LS患者丘脑、基底节、脑室、大脑导水管周围和脊髓后柱区有对称性局灶坏死,呈海绵状囊样空腔,并伴有脱髓鞘改变、血管增生和神经胶质增生[14]。LS具有复杂的临床表现和基因调控机制,为严重的致死性遗传病,且目前无有效治疗措施,故产前诊断是一项重要预防措施。
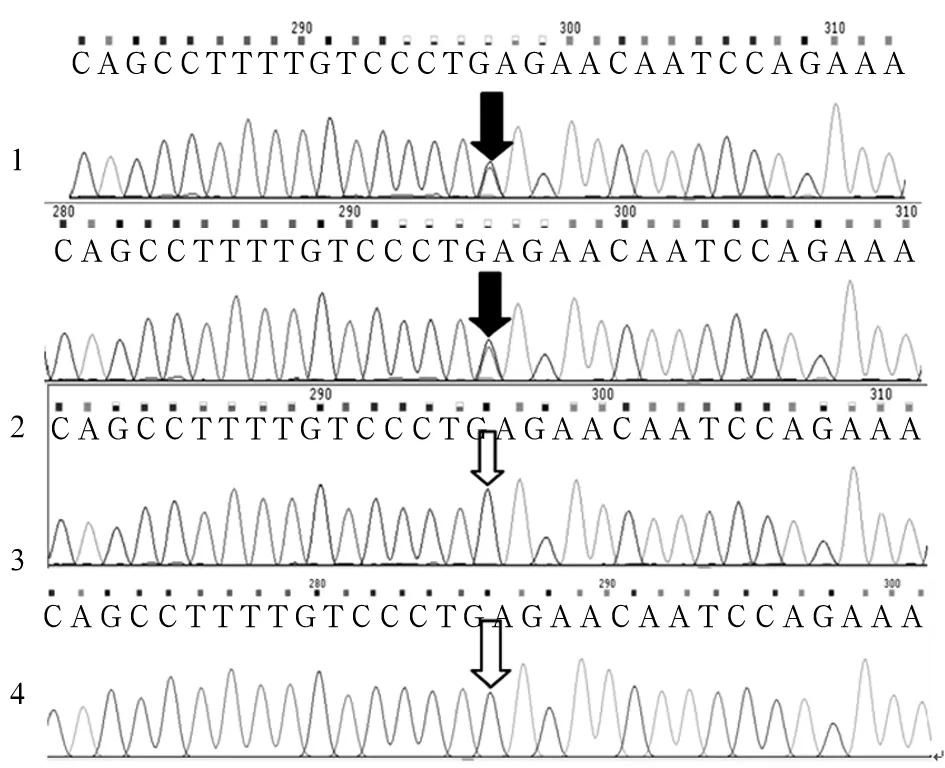
注:1为患儿样本;2为患儿父亲样本;3为患儿母亲样本;4为羊水细胞样本;黑色箭头示SURF1基因存在突变位点c.324-1G>C,c.655G>T;白色箭头处SURF1基因不存在突变位点c.324-1G>C,c.655G>T。
图2 SURF1基因突变位点c.324-1G>C,c.655G>T
LS主要有3种遗传方式:线粒体遗传(又称母系遗传),由线粒体基因突变引起;常染色体隐性遗传;X-连锁遗传。后两种遗传方式是核基因突变所致,其中常染色体隐性遗传最为常见,约占60%。目前发现约10种类型核基因突变引起线粒体病,包括呼吸链复合物亚基及组装因子突变,维持线粒体基因稳定性的突变,线粒体蛋白组合装置缺陷,线粒体脂质环境缺陷,线粒体运输装置缺陷,辅酶Q10缺乏,线粒体网络动态学缺陷,凋亡缺陷,丙酮酸脱氢酶缺陷和线粒体铁代谢障碍[3]。呼吸链复合物亚基及组装因子突变基因达51个[15]。研究[16,17]发现常染色体隐性遗传与4种呼吸链酶缺陷有关,即复合物Ⅰ(NADH还原酶)、复合物Ⅱ(琥珀酸脱氢酶)、复合物Ⅳ(COX)、复合物Ⅴ(ATP合成酶)。其中最为常见是复合物Ⅳ的缺陷。COX复合物由13个多肽亚单位组成,其中10个由核基因编码(SURF1、SCO1、SCO2、COX10、COX15、COX20、LRPPRC、FASTKD2、TACO1、PET100等),其余3个由线粒体基因编码[18,19]。LS最常见的致病基因是SURF1,位于9q34.2,有9个外显子,全长4 695 bp。SURF1基因编码的蛋白含有300个氨基酸残基(AA),有2个跨膜结构域(61~79AA、274~290AA)。SURF1蛋白在COX复合物的装配前期中发挥十分重要的作用。迄今国外报道已发现40多例LS患者存在SURF1基因突变[20];国内共报道了6例,1例为622delA和653 -654delCT缺失,5例为外显子7的604位G>C杂合错义突变。
本研究中患儿发育落后,不能独走,扶走时双下肢抬起困难,2岁后活动能力有倒退;血乳酸3.0 mmol/L;头部MRI检查示双侧基底节区及脑干异常信号,部分病变弥散受限,符合LS的临床特征,拟诊断为LS。进行二代测序筛查基因发现患儿SURF1基因有c.655G>T(p.E219X)及c.324-1G>C复合杂合突变,与PubMed文献数据库、美国生物技术信息中心(NCBI)所属的SNP数据库、人类基因突变数据库(HGMD)中已公布的SURF1基因突变位点相比,这两个突变均未见报道。上述两个突变中,前者为无义突变,后者为剪切突变,理论上均具有致病性。故我们认为这两种缺失突变为新的致病突变。传递分析表明,患儿母亲存在SURF1基因c.655G>T杂合突变,父亲有SURF1基因c.324-1G>C杂合突变,明确了诊断和致病基因,不仅为患儿的治疗提供了准确依据,而且根据患儿致病SURF1基因及其父母基因比对,发现患儿父母亲均为致病基因携带者,遗传方式是常染色体隐性遗传,故患儿父母再次生育时,其子女患病概率为25%,需进行产前诊断。本例患儿母亲在孕18周时进行羊水细胞PCR检测,发现胎儿有c.324-1G>C突变,没有c.655G>T突变,为杂合突变携带者,理论上不具致病性,故保留此胎儿,产后随访至1岁,孩子未出现异常。
[1] 李欣,曹翔,汪麟,等.疑难病例分析:线粒体脑病1例[J].齐齐哈尔医学院学报,2015,36(13):2027-2028.
[2] Graham BH. Diagnostic challenges of mitochondrial disorders:complexities of two genomes[J]. Methods Mol Biol, 2012,837:35-46.
[3] 刘志梅,方方.核基因突变导致儿童线粒体病的分子遗传学进展[J].中国循证儿科杂志,2015,10(6):470-474.
[4] Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children[J]. Neurology, 2006,67(10):1823-1826.
[5] Leigh D. Subacute necrotizing encephalomyelopathy in an infant[J]. Neurol Neurosurg Ychiatry, 1951,14:216-221.
[6] 刘雪梅,魏晓晶,苏飞飞,等.Leigh综合征1例报告[J].中风与神经疾病杂志,2017,34(2):177-178.
[7] DiMauro S, Schon EA. Mitochondrial disorders in the nervous system[J]. Annu Rev Neurosci, 2008,31(10):91-123.
[8] Scheffer RC, Smout AJ. Tachyduodenia in mitochondrial neurogastrointestinal encephalomyopathy[J]. Neurogastroenterol Motil, 2011,23(5):408-410.
[9] 常凯杰,马明明,张晓辉,等.肢带型线粒体肌病一个家系的临床和病理特点研究[J].国际神经病学神经外科学杂志,2016,43(1):22-26.
[10] 王丽辉,郑华城,杨花芳,等.儿童Leigh综合征4例临床分析[J].临床儿科杂志,2016,34(2):111-114.
[11] Ruhoy IS, Saneto RP. The genetics of Leigh syndrome and its implications for clinical practice and risk management[J]. Appl Clin Genet, 2014,7(12):221-234.
[12] 孟庆林,江炜炜.线粒体脑肌病伴高乳酸血症和卒中样发作综合征的临床特点分析[J].临床神经病学杂志,2016,29(4):274-277.
[13] 祝小芬,丁冀,朱筱琦,等.线粒体肌病临床病理学特征及简易乳酸运动试验的筛选价值[J].中国现代神经疾病杂志,2016,16(12):859-864.
[14] 卢万协.婴幼儿Leigh综合征的MRI诊断探讨分析[J].河南预防医学杂志,2009,20(5):402-403.
[15] Chinnery PF, Hudson G. Mitochondrial genetics[J]. Br Med Bull, 2013,106(5):135-159.
[16] Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing[J]. Med Genet, 2012,49(4):277-283.
[17] Nogueira C, Barros J, Sa MJ, et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency[J]. Neurogenetics, 2013,14(2):153-160.
[18] Bohm M, Pronicka E, Karczmarewicz E, et al. Retrospective, Multicentric Study of 180 Children with Cytochrome c Oxidase Deficiency[J]. Pediatr Res, 2006,59(4):21-26.
[19] Szklarczyk R, Wanschers BF, Nijtmans LG, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia[J]. Hum Mol Genet, 2013,22(4):656-667.
[20] Zhang Y, Sun F, Kong QP, et al. Mutation analysis on nuclear gene and mitochondrial gene for Chinese patients with Leigh syndrome[J]. Clin Pediatr, 2008,26(12):1021-1025.
10.3969/j.issn.1002-266X.2017.27.028
R394
B
1002-266X(2017)27-0090-03
2017-03-05)
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
种子(2021年3期)2021-04-12 01:42:22
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
中国生殖健康(2020年2期)2021-01-18 02:51:26
中成药(2018年7期)2018-08-04 06:04:18
小学生导刊(2018年13期)2018-06-29 03:49:00
中成药(2018年3期)2018-05-07 13:34:18
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
河南医学研究(2014年5期)2014-02-27 14:52:41
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
