
CH3I2+的二体、三体解离过程的理论研究∗
2017-08-09 00:32孙启响闫冰
物理学报 2017年9期
孙启响 闫冰
1)(吉林农业大学信息技术学院,长春 130118)2)(吉林省应用原子分子光谱重点实验室(吉林大学),吉林大学原子与分子物理研究所,长春 130012)
CH3I2+的二体、三体解离过程的理论研究∗
孙启响1)2)闫冰2)†
1)(吉林农业大学信息技术学院,长春 130118)2)(吉林省应用原子分子光谱重点实验室(吉林大学),吉林大学原子与分子物理研究所,长春 130012)
(2016年10月31日收到;2017年2月1日收到修改稿)
采用密度泛函和耦合簇理论从过渡态的观点对CH3I2+离子的解离过程进行了计算和分析.优化得到了CH3I,CH3I+和CH3I2+的稳定结构及解离过渡态的几何构型并给出了相应能量,计算的第一、二电离能与实验结果符合.基于母体离子、过渡态和解离碎片的几何结构和能量,对CH3I2+的两体解离过程和三体解离过程进行了详细分析和讨论.给出了二体、三体解离的可能解离通道,并分析了实验上观测到的解离碎片离子的产生机制.计算结果表明,三重和单重绝热势能面上的一些解离过程表现出明显的差异.
密度泛函理论,过渡态,二体/三体解离,CH3I2+
1 引 言
碘化学激光器是一种新型高能化学激光器,有着激光波长更短、效率更高、光束质量较好、储能高、装置更安全简便等优点,广泛应用于工业和军事领域.碘代烷烃解离产生第一激发态的碘原子可用作碘激光的工作物质.碘原子基态I(2P3/2)和第一激发态I∗(2P1/2)间的磁偶极跃迁产生波长为1.315µm的激光,已用于石英光纤的高效传输[1].而碘甲烷CH3I作为基础化学反应的典型分子,其电离解离过程一直是人们研究的热点[2−36].
在实验方面,近年的研究主要集中在CH3I在强激光场中的电离解离过程.2001年,Lehr等[15]用强度为1016W.cm−2,50 fs激光脉冲作用于CH3I分子上,观测到了母体离子CH3I+和CH3I2+及解离碎片I的1—7价的阳离子,H+,Cp+(p≤ 4)等产物.Locht等[31]通过对碘甲烷分子的光致解离光谱测定得知,I+,CH2I+等产物集中出现在12.2—12.7 eV的光子能量区间内,并指出这三种离子是由基态X2E的解离和Rydberg态的自电离解离产生的.355 nm激光作用于CH3I分子,在飞行时间质谱中发现了信号较强的H+,和I+,同时也观测到信号较弱的C+,CH+,和CH3I+产物[32].可见,实验上观测到了稳定的一价母体离子CH3I+和二价离子CH3I2+,也观测到了其他各种碎片离子,包括I+,以及其他碳氢碎片离子.因此,给出实验中观测到的各种碘甲烷碎片离子的电离解离通道是当前理论计算研究的一个重要任务.
在理论计算方面,针对CH3I分子的电子结构和电离解离的研究已有诸多报道[29−36].Alekseyev等[35]率先研究了包含自旋-轨道耦合效应的CH3I分子沿C—I键解离的基态和激发态的势能曲线.对于中性分子,前人的研究主要集中在C—I解离势能曲线交叉和各激发态对吸收光谱的贡献[2,34,35].Adjeddine等[36]采用组态相互作用方法研究了CH3I+离子电子基态和激发态,计算了与1S)四个解离限对应的18个电子态的势能曲线,并获得了束缚态的解离能和几何结构信息.
综上所述,CH3Iq+(q=0,1,2)体系的解离动力学已引起广泛的实验和理论研究兴趣,但主要集中在中性和一价离子,而对于实验上观测到的二价CH3I2+离子的解离/电离的理论研究较少.本文基于过渡态的观点对于CH3I2+离子的二体、三体解离进行了理论研究,计算结果可进一步加深对激光场中CH3I的电离解离实验的理解.
2 计算方法
基于密度泛函与耦合簇理论对CH3I2+的解离程进行了理论计算与分析.C,H两原子采用aug-cc-pVTZ基组[37],为了克服多电子数目给计算带来的困难,我们采用了引入赝势基组的冻结芯近似处理方法,I原子采用赝势ECP46 MWB[38]及 相 应 的AVTZ[39](15s11p4d2f)/[4s4p3d2f]收 缩基组来描述价电子.在Cs分子对称性下优化得到了CH3I分子及其离子CH3I+,CH3I2+的几何结构,以及CH3I2+的各种异构体及过渡态;对于二价离子CH3I2+,计算研究了最低的单重态(1A′)和三重态(3A′′),以得到在两个电子态绝热势能面上的解离信息.首先,我们采用密度泛函理论(DFT)在B3LYP/6-311G(d,p)水平上优化得到解离过程中的所涉及的异构体及过渡态的几何结构,并在相同水平上计算了谐振频率,获得各结构相应的零点振动能(ZPE)修正;旋轨耦合相对论修正能量(Esoc)是基于优化的几何结构,采用包含自旋-轨道耦合赝势的基组计算得到的.优化得到的同分异构体的振动频率都为正,而过渡态结构至少有一个虚频.为准确考虑电子相关效应,我们在DFT优化的几何结构基础上,采用耦合簇理论CCSD(T)和相同的基组计算了CH3I2+的异构体及过渡态结构的单点能.最后,对CH3I2+的二体、三体解离动力学过程中涉及碎片进行了相同水平的计算.
3 结果与讨论
3.1几何结构计算
我们采用DFT-B3LYP/6-311G(d,p)方法计算了中性和一价CH3I体系的几何结构以及二价离子CH3I2+的二体和三体解离过程可能包含的异构体、过渡态和解离碎片的结构.中性碘甲烷分子的对称性为C3v,为了避免其离子可能的对称性降低,CH3Iq+体系的电子结构计算是在C3v的阿贝尔子群Cs或C1下进行的,计算获得的几何构型如图1所示.表1列出了CH3Iq+体系及一些可能的离子结构在稳定点的能量,其中也包含了相同理论水平下计算的ZPE修正,自旋-轨道耦合相对论修正Esoc及各结构对应的单点能(EDFT和ECCSD(T)),其中EDFT表示DFT计算的能量,ECCSD(T)表示耦合簇理论计算的单点能.表中常数Na表示该结构对应的虚频个数;能量E表示DFT计算所得能量EDFT与ZPE之和;E1为耦合簇理论结果与ZPE修正之和;E2为耦合簇理论结果与ZPE修正及自旋-轨道耦合相对论修正之和.能量零点取为相同理论水平和修正下的中性碘甲烷分子在平衡几何结构下的能量.如图1中(1)构型所示,CH3I分子基态的主要几何结构参数为RC—H=1.082 Å,RC—I=2.162 Å,∠H—C—H=107.3◦,与组态相互作用(confuguration interaction,CI)计算结果(RC—H=1.084 Å,RC—I=2.132 Å, ∠H—C—H=111.20◦)[27]和实验结果(RC—H=1.084 Å,RC—I=2.1358 Å,∠H—C—H=111.4◦)[28]相比,计算结果基本符合;图1中的结构(2)为CH3I+离子的基态结构,其主要几何参数 (RC—H=1.082 Å,RC—I=2.158 Å,∠H—C—I=107.2◦),与CASSCF计算结果(RC—H=1.0721 Å,RC—I=2.2082 Å,∠H—C—I=107.00◦)近似,最大差异为C—I键长相差0.05 Å.在优化得到的CH3I2+离子的24种同分异构中,共有4个局域极小值稳定结构,分别为(3a),(3b),(8a)和(8b);其中(3a)和(3b)构型是CH3I2+离子1A′态和3A′′态,在Cs对称性下的稳定结构(8a)和(8b)分别为这两个电子态在C1对称性下的异构体.(8a)构型所对应的电离能量最低,为26.01 eV.在所有的过渡结构中,能够发生两体解离的构型有13种,直接发生三体解离过程的过渡态结构为(5)和(7)构型.我们计算的CH3I分子第一垂直电离能9.4 eV与实验值9.54 eV[22]符合较好.CH3I2+(1A′)和CH3I2+(3A′′)的垂直电离能分别为29.17 eV和27.69 eV,考虑电子相关、旋-轨耦合相对论效应和ZPE修正之后分别为28.80 eV和27.40 eV,与CI计算结果(29.6±0.3)和(27±0.3)eV[25]接近,差异分别为0.5 eV,0.1 eV;第二垂直电离能27.40 eV与实验值26.7 eV[33]接近.

表1 CH3I分子及其离子CH3I+,CH3I2+的虚频个数及相关能量Table 1.The number of imaginary vibrational frequencies and the corresponding energies for isomers of neutral CH3I,CH3I+cation,and CH3I2+dication.
3.2CH3I2+离子两体和三体解离
实验上,Kaziannist等[27]将35 ps激光场与CH3I分子相互作用,在质谱中观测到和I+离子有着较高的量子产额,并给出了可能的通道:CH3I2+发生两体解离生成我们计算研究了CH3I2+离子的最低单重态和三重态的两体解离及后续的三体解离过程,相应的绝热解离势能面可分别用如下两个反应式表示:


图1 优化的CH3I分子及其离子CH3I+和CH3I2+的几何结构,包括CH3I2+离子4种异构体和20种过渡态结构(键长:Å;键角:(◦))Fig.1.The optimized geometries for the neutral CH3I molecule,CH3I+cation,and CH3I2+dication including 4 isomers and 20 transition states of CH3I2+(bond length: Å;bond angle:(◦)).
其中(1)式为单重态势能面上的解离,(2)式表示三重态解离,ts表示过渡态,上述两式中所标记的数字为中间过渡态的E2能量.由(1)和(2)式可见单、三重绝热势能面发生解离会产生不同电子态的I+离子.
图2中,垂直线表示中性CH3I分子垂直电离生成可能的二价阳离子CH3I2+,实线和虚线分别代表单重态和三重态,其中,三重态的能量较低,对称性为C2v,电子态为3A2;单重态为Cs对称性下的1A′态.中性碘甲烷分子垂直电离生成单重态二价离子CH3I2+(1A′),二次垂直电离能约为29 eV,之后,二价离子会发生几何结构弛豫,主要表现为∠H—C—H和∠H—C—I的变化,弛豫到能量局域极小值点(3a),再经过相对能量为27.78 eV的过渡态结构(4a)∠H—C—H=119.1◦,∠H—C—I=94.0◦),即越过高为0.3 eV的势垒实现的二体解离;值得注意的是,此二体解离是一个放能过程,释放能量约为3.5 eV.在本文中,为了与电离解离实验比较,吸收或释放能量均为与相应的垂直电离能比较而言的.
经垂直电离生成的相对能量较低的三重态CH3I2+(3A2)二价离子经微小变化(∠H—C—H增 大2.9◦, ∠H—C—I减 小3.4◦), 可 弛 豫 到 稳定结构(3b);在经过渡态结构(4b)(RC—H=1.090 Å,RC—I=2.797 Å,∠H—C—H=119.5◦,∠H—C—I=94.1◦)实现的二体解离,此过程只需克服约0.1 eV的势垒;与单重态解离(1)式类似,三重态的垂直电离到二体解离过程同样是一个放能过程,释放能量约2 eV.进一步地,经过渡结构,最终实现三体解离即反应(2)式.
综上所述,生成三重态的CH3I2+所需电离能更低,在实验上更易于生成.依据考虑修正后计算的E2结果,无论三重态还是单重态,与垂直电离生成二价离子所需能量相比,其二体解离均为放能过程,释放能量为几电子伏量级;进一步地,三体解离的阈值略低于垂直电离能,可见(1)和(2)式中所示解离过程均可经由二次垂直电离自发生成,并不需要吸收额外能量即可实现.即单重态解离(1)式所需能量为28.89 eV,三重态(2)式需要最小能量为27.60 eV.我们计算的能量阈值与Dujardin等[28]实验测得所需能量(28.5±0.5)eV接近,也与实验值(27.0±0.3)eV[27]符合较好.
碘甲烷分子经垂直电离并弛豫到稳定的单重和三重电子态后,会发生不同的二体解离,并进一步发生三体解离生成不同的碎片.结合图3所示的解离势能曲面,(3)式描述了稳定的单重二价离子(3a)吸收约5 eV能量,经过一个能量为32.58 eV的过渡结构(ts5)(RC—H1=3.308 Å,RC—H2=1.620 Å,RC—H3=2.328 Å,RC—I=1.905 Å,∠H1—C—H2=90.0◦,∠H1—C—I=150.6◦),发生库仑爆炸,直接生成CI+(1Σ+)+H2+H+的三体解离过程.而对于三重态的稳定二价离子(3b),其解离过程与单重态离子(3a)表现出明显的差异.
(4)—(6)式描述了能量较低的三重态二价碘甲烷离子发生非电荷对称的二体解离,进而生成三体解离产物的物理过程.由图3可知,在(4)—(6)式中三重稳定结构(3b)需要吸收额外的约3 eV的能量,经过过渡结构(ts6)(RC—H1=3.224 Å,RC—H2=1.095 Å,RC—H3=1.095 Å,RC—I=1.966 Å, ∠I—C—H2=118.9◦, ∠H3—C—I=125.2◦),发生非对称解离生成CH2I2+和H原子;之后,额外的2—3 eV能量可进一步使CH2I2+发生C—I,C—H断裂,其中对应(6)式的C—I断裂的电荷对称解离产物能量最低.总体看来,(3)—(6)式描述的二体、三体解离均为吸能过程,垂直电离的碘甲烷二价离子需要额外吸收4—5 eV能量才可发生上述解离过程;其中单重态直接解离(3)式阈值为32.58 eV,(4),(5)和(6)式三体解离发生的阈值分别为32.56和31.26 eV.
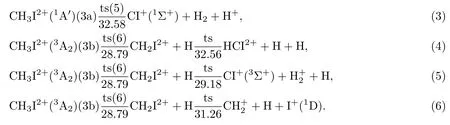

图3 CH3I2+→CI+(1Σ+)+H2+H+等解离过程的势能面Fig.3.The potential energy surfaces for the CH3I2+→CI+(1Σ+)+H2+H+dissociation process.
3.2.3 CH3I2+→CI++H2+H+/HCI++H+H+
图4和(7)—(8)式 描 述CH3I2+→CI++H2+H+/HCI++H+H+解离过程及相关能量.CH3I2+离子稳定结构(3a)和(3b)分别经过渡态结构(13a)和(13b),生成稳定的结构(8a)和(8b),上述过程实质为氢原子/质子转移过程,CH3上的一个H转移到I原子上(见图1);然后,发生电荷对称二体解离,产生CH3I2+→CH2I++H+;CH2I+进一步解离,最终单重和三重态碘甲烷二价离子分别生成CI++H2+H+和HCI++H+H+,该过程分别对应(7)和(8)式. 由图4可知,单重态三体解离(7)式的阈值为28.89 eV,不需要吸收额外能量;三重态解离(8)式的阈值为29.25 eV,需要吸收额外能量约1.5 eV.Dujardin等[28]在对CH3I分子进行的PIPICO实验中测得反应CH3I+hv→CH3I2+→CH2I++H+的阈值能量为(31.0±0.5)eV,由图4可知,对于实验中更容易生成的能量较低的三重态CH3I2+离子,其二体解离生成CH2I++H+的阈值能量为翻越过渡态(9b)需要的能量,为29.25 eV,与实验值[28]的下限十分接近.


图4CH3I2+→CI++H2+H+/HCI++H+H+解离势能面Fig.4.The potential energy surfaces for the CH3I2+→CI++H2+H+/HCI++H+H+dissociation process.
图5和(9)—(11)式中的二体解离产生了氢分子或氢分子离子,其中单重态对应的(9)和(10)式为对称解离,生成了,而三重态对应的(11)式为非对称解离,产生了中性氢分子.如图5所示,非对称解离产物的能量较高,这符合一般的库仑爆炸反应规律;单重态发生的二体解离为放热反应,而三重态与此相反,为一吸热过程,这与实验[14]中所得解离产物中的产率比H2高的结果相符合.进一步的解离中,(9)和(10)式中分别发生了的碎裂和HCI+的氢原子解离,而后者需要的能量较低,(9)和(10)式三体解离的阈值分别为30.62 eV和28.89 eV,其中前者为吸热过程,后者为放热过程;(11)式对应的最终生成了中性的氢分子、质子和CI+离子,是阈值最高的解离过程,为33.41 eV.
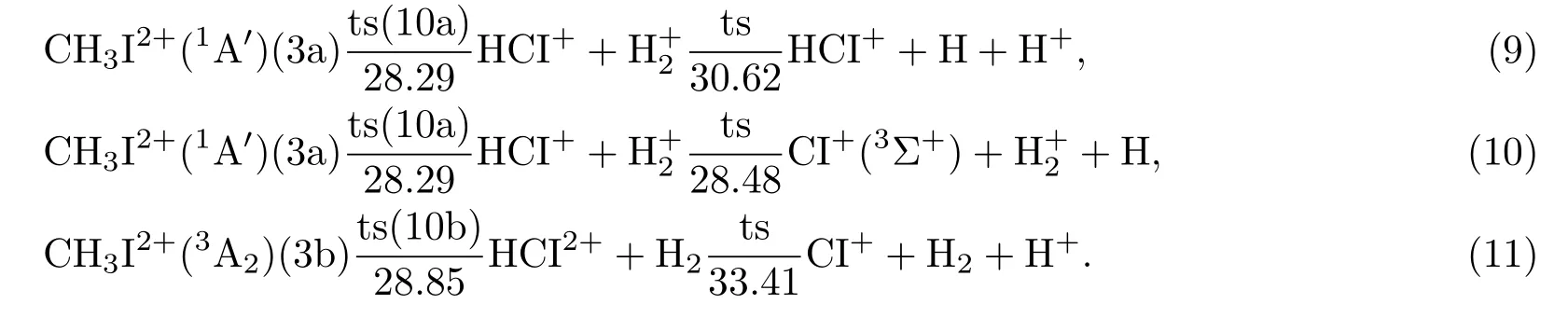

图5CH3I2+发生解离的势能面Fig.5.The potential energy surfaces for thedissociation process.
在图6以及(12)和(13)式中,无论单、三重态,二体解离均产生了离子,而进一步的反应也都是碎裂为氢分子离子和氢原子的过程.上述的三体解离均为吸热过程,并且单重态解离的阈值低于三重态的阈值,分别为29.55 eV和31.87 eV.


图6CH3I2+解离为的势能曲面Fig.6.The potential energy surfaces for thedissociation process.
在实验中,Lehr等[15]观测到了的产生,本节的计算给出了可能的产生通道.在图7以及(14)和(15)式中,由二次电离到生成稳定的CH2IH2+结构(8a)和(8b)过程,单、三重态均与3.2.3节中过程相同;之后的二体解离生成了亚甲基离子和碘化氢离子,从中性分子二次电离获得的能量来看,其中单重态生成亚甲基离子不需要额外吸收能量,而三重态为吸能过程.进一步,三体解离生成了亚甲基离子、碘离子和氢原子.三体解离过程,单重态和三重态的解离行为是一致的,区别在于产生了不同自旋多重度的碘离子.单重态(14)式的三体解离能量阈值为28.89 eV,是放热过程;三重态(14)式的三体解离阈值为28.88 eV,是吸热过程.
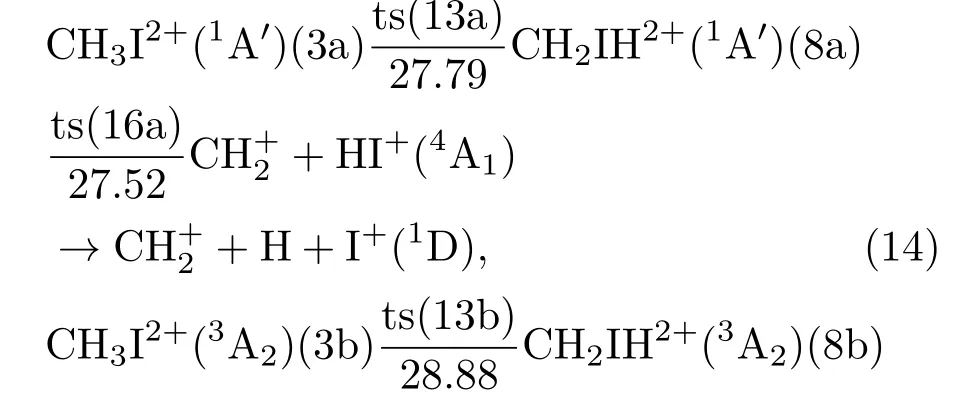

综上所述,我们计算了碘甲烷中性分子经二次电离生成的能量最低的单重和三重CH3I2+离子,对应的垂直电离能分别为28.89 eV和27.6 eV.在表2和表3中,我们总结了由垂直电离产生的二价离子,不再吸收额外能量而自发解离的二体、三体通道及对应的碎片信息;由于不再额外吸收能量,在激光与碘甲烷相互作用的实验中较易观测到这些最可能解离通道产生的解离碎片.

图7CH3I2+发生CH+2+H+I+解离的能量图Fig.7.The potential energy surfaces for theCH3I2+→CH+2+H+I+dissociation process.
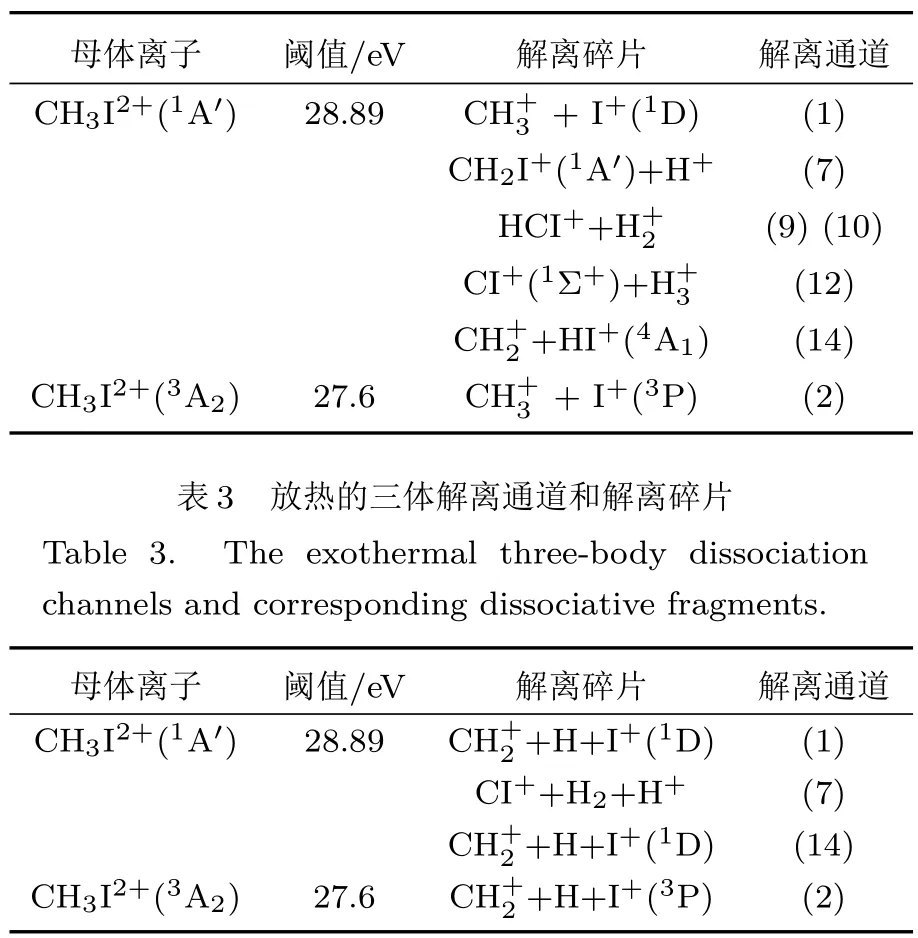
表2 放热的二体解离通道和解离碎片Table 2.The exothermal two-body dissociation channels and corresponding dissociative fragments.
4 结 论
本文对CH3I2+离子的解离通道进行了计算和分析.优化得到了CH3I,CH3I+和CH3I2+的稳定结构及解离过渡态的几何构型并给出了相应能量,计算的第一、二电离能与实验结果符合;计算发现,CH3I2+的基态为三重态3A2.对CH3I2+的11种两体解离过程和15种三体解离过程进行了详细分析和讨论.对于CH3I2+离子,我们仅从能量的角度总结了解离过程实现的难易程度.上述所有的11种二体解离过程中,除相应的二次电离能外,不需要额外吸收能量即可发生的解离有单重态CH3I2+(1A′)解离生成三重态的解离产生这些放能解离通道应为质谱实验[34]中观测到的信号较强的离子的可能产生通道;给出了其他离子(例如)的可能产生通道,需要进一步的实验验证.同时,我们也给出了几种除二次电离外不需要额外吸收能量的三体解离通道,这些通道会产生亚甲基离子、碘离子、氢离子、氢分子离子等.在上述过程的计算中,我们发现DFT和CCSD(T)给出的单点能量计算结果有时会有差异,本文的结果是以后者的计算结果为主进行讨论的.我们的计算结果也表明三重和单重势能面上的解离过程表现出较大差异性.
[1]Cheng L 2007Ph.D.Dissertation(Harbin:Harbin Institute of Technology)(in Chinese)[程丽 2007博士学位论文(哈尔滨:哈尔滨工业大学)]
[2]Li R,Yan B,Zhao S T,Guo Q Q,Lian K Y,Tian C J,Pan S F 2008Acta Phys.Sin.57 4130(in Chinese)[李瑞,闫冰,赵书涛,郭庆群,连科研,田传进,潘守甫2008物理学报57 4130]
[3]Liu H T,Yang Z,Gao Z J 2007J.Chem.Phys.126 044316
[4]Goss S P,Mcgilvery D C,Morrison J D,Smith d L 1981J.Chem.Phys.75 757
[5]Schneider W,Thiel W 1989Chem.Phys.Lett.157 367
[6]Continetti R E,Balko B A,Lee Y T 1988J.Chem.Phys.89 3383
[7]Mintz D M,Baer T 1976J.Chem.Phys.65 2407
[8]Lee M,Kima M S 2007J.Chem.Phys.127 124313
[9]Sparks R K,Shobotake K,Carlson L R,Lee Y T 1981J.Chem.Phys.75 3838
[10]Zhong D,Cheng P Y,Zewail A H 1996J.Chem.Phys.105 7864
[11]Imre D,KinseY J L,Sinha A,Krenos J 1988J.Phys.Chem.89 6667
[12]Sundberg R L,Imre D,Hale M O,Kinsey J L,Coalson R D 1986J.Phys.Chem.90 5001
[13]Johnson B R,Kittrell C,Kelly P B,KinseY J L 1996J.Phys.Chem.100 7743
[14]Amatatsu Y,Yabushita S,Morokuma K 1996J.Chem.Phys.104 9783
[15]Lehr L,Weinkauf R,Schlag E W 2001Int.J.Mass.Spectrom.206 191
[16]Walter K,Weinkauf R,Boesl U,Schlag E W 1988J.Chem.Phys.89 1914
[17]Sharma P,Vatsa R K,Rajasekhar B N,Das N C,Ghanty T K,Kulshreshtha S K 2005Rapid Commun.Mass.Spectrom19 1522
[18]Zhang B L,Wang X Y,Lou N Q,Zhang B 2001J.Spec.Acta Part A57 1759
[19]Chupka W A,Colson S D,Seaver M S,Wooddard A M 1983Chem.Phys.Lett.95 171
[20]Shapiro M,Bersohn R 1980J.Chem.Phys.73 3810
[21]Karlsson L,Jadmy R,Mattson L,Chau F T,Siegbahn K 1977Phys.Scripta16 225
[22]Landolt H,Börnstein R,Fischer H,Madelung O,Deuschle G 1987Landolt-Bornstein:Numerical Data and Functional Relationships in Science and Technology(Vol.17)(Berlin Heidelberg:Springer-Verlag)
[23]Ragle J L,Stenhouse I A,Frost D C,Mcdowell C A 1970J.Chem.Phys.53 178
[24]Randic M,Trinajstic N 1992J.Chem.Edu.69 701
[25]Griffiths W J,Harris F M,Parry D E 1990J.Chem.Soc.Faraday Trans.86 2801
[26]Yabushita S,Morokuma K 1988Chem.Phys.Lett.153 517
[27]Kaziannis S,Siozos P,Kosmidis C 2005Chem.Phys.Lett.401 115
[28]Dujardin G,Hellner L,Winkoun D,Besnard M 1986J.Chem.Phys.105 291
[29]Guo H 1992J.Chem.Phys.96 2731
[30]Roth J,Tsitrone E,Loarer T,Philipps V,Brezinsek S,Loarte A 2008Plasma Phys.Control.Fusion50 103001
[31]Locht R,Dehareng D,Hottomann K,Kaziannis H,Jochims W,Hbaumgartel L B 2010J.Phys.B:At.Mol.Opt.Phys.43 105101
[32]Li L,Kong X H,Zhang S D,Liu C H,Sun Z Q,Liu J P,Zhang L F,Qiao G 2007J.Atom.Mol.Phys.3 443(in Chinese)[李丽,孔祥和,张树东,刘存海,孙志青,刘建苹,张良芳,乔光2007原子与分子物理学报3 443]
[33]Griffiths W J,Franck M,Harris,Parry D E 1990J.Chem.Soc.Faraday Trans.86 2801
[34]Ajitha D,Wierzbowska M,Lindh R,Malmqvist P A 2004J.Chem.Phys.121 5761
[35]AlekseyevAB,LiebermannH P,BuenkerR J,Yurchenko S N 2007J.Chem.Phys126 234102
[36]Adjeddine M,Flament J P,Teichteil C 1987Chem.Phys.118 45
[37]Dunning T H 1989J.Chem.Phys.90 1007
[38]Bergner A,Dolg M,Kuechle W,Stoll H,Preuss H 1993Mol.Phys.80 1431
[39]Martin J M L,Sundermann A 2001J.Chem.Phys.114 3408
PACS:31.15.aj,31.15.E–,31.50.–xDOI:10.7498/aps.66.093101
Computational study of two-body and three-body dissociation of CH3I2+∗
Sun Qi-Xiang1)2)Yan Bing2)†
1)(Faculty of Information Technology,Jilin Agricultural University,Changchun 130118,China)2)(Jilin Provincial Key Laboratory of Applied Atomic and Molecular Spectroscopy,Institute of Atomic and Molecular Physics,Jilin University,Changchun 130012,China)
31 October 2016;revised manuscript
1 February 2017)
As one of the simplest alkyl halides,methyl iodide is extensively investigated in the research fi elds of the photodissociation and photoionization.In the experimental investigations of ionization and dissociation,many molecular fragments,such as Iq+(q≤3),,H+,etc.,are observed in the mass spectrum of CH3I.While the mechanisms for dissociation and ionization are not completely understood.As the doubly-ionized product,CH3I2+exhibits di ff erent isomer structures and isomerization reactions.The dissociation channels of di ff erent isomers in combination with the corresponding transition states of CH3I2+are helpful for better understanding the dissociation and ionization dynamics of CH3I in an intense laser fi eld.
In our present work,the dissociation channels of CH3I2+are investigated by the density functional and couple cluster theory.The geometries and energies corresponding to the local isomers and the transition states of CH3I,CH3I+and CH3I2+are computed.The fi rst and second ionization energies we measured are in good agreement with experimental values.Our computational results show that the ground state of the CH3I2+is a triplet one with3A2symmetry.Totally 11 two-body and 15 three-body dissociation channels of the CH3I2+on both the lowest singlet and the lowest triplet potential energy surfaces are computed and analyzed in detail.Our computations indicate that seven two-body dissociations channels,i.e.,six singlet and one triplet ones,are exergonic,in whichis the easiest process to achieve;four exergonic three-body dissociation channels with three on singlet potential energy surface and one on triplet potential energy surface are found.The possible mechanisms for producing the dissociative ionized fragments observed in experiments,and I+,are presented;furthermore,the dissociation channels generating other ions not observed in experiments,such aset al,are also given for further experimental study.The detailed information about dissociation channels and fragments is summarized for further experimental comparisons.In the computations,we fi nd that the density functional theory and CCSD(T)methods give di ff erent energy orders for a few dissociation potential energy surfaces;and in this work,the analysis and discussion are performed based on the CCSD(T)results.Our computations indicate that the dissociation channels on singlet and triplet potential energy surface exhibit di ff erent behaviors.
density functional theory,transition states,two-body and three-body dissociation,CH3I2+
10.7498/aps.66.093101
∗国家自然科学基金(批准号:11574114)、吉林省自然科学基金(批准号:20150101003JC)和吉林农业大学科研启动基金资助的课题.
†通信作者.E-mail:yanbing@jlu.edu.cn
*Project supported by the National Natural Science Foundation of China(Grant No.11574114),the Natural Science Foundation of Jilin Province,China(Grant No.20150101003JC),and the Scienti fi c Research Foundation of Jilin Agricultural University,China.
†Corresponding author.E-mail:yanbing@jlu.edu.cn
猜你喜欢
广东药科大学学报(2022年5期)2022-12-30
北京航空航天大学学报(2022年5期)2022-06-06
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中老年保健(2019年9期)2019-01-13
电脑知识与技术(2018年3期)2018-03-21
中学教学参考·理科版(2016年3期)2017-05-19
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
中学化学(2015年10期)2015-12-14
中国有色冶金(2015年5期)2015-03-06
