
氯法齐明在大鼠体内的药物代谢动力学及组织分布研究
2017-08-07 06:56付寒杨春燕王俊张小平杨应周
中国防痨杂志 2017年8期
付寒 杨春燕 王俊 张小平 杨应周
·论著·
氯法齐明在大鼠体内的药物代谢动力学及组织分布研究
付寒 杨春燕 王俊 张小平 杨应周
目的 研究氯法齐明在正常大鼠体内的药物代谢动力学及组织分布特征。方法 将平均体质量(200±20) g的Sprague-Dawley(SD)雄性大鼠按随机数字表法分成8组,每组5只。分为单次给药实验组包括空白对照组(BL)、空腹实验组(KF)、普通饮食组(NM)、高脂饮食组(HM)、低剂量组(NL)、高剂量组(NH)、中剂量组(即普通饮食组NM);多次给药实验组包括空白对照组(D0)和多次给药组(D1)。单次给药实验组中低、高剂量组给药剂量分别为2.1 mg/d、12.6 mg/d,其他组均为中剂量6.3 mg/d;多次给药组剂量为2.1 mg/d。空白对照组(BL组和D0组)均以油溶剂代替药物溶液。给药后不同时间点取血及组织样本,应用液相色谱-质谱联用法(LC-MS/MS)测定氯法齐明在大鼠血浆和组织液中的药物浓度,并计算药物代谢动力学参数血药浓度-时间曲线下面积(AUC) 、峰浓度(Cmax) 、达峰时间(Tmax) 和半衰期(T1/2)。结果 氯法齐明单次给药后,Cmax值HM组为(1348.05±208.46) ng/ml,明显高于NM组[(719.33±123.70) ng/ml;t=5.77,P<0.01]和KF组[(557.73±85.11) ng/ml;t=2.41,P<0.01];Tmax值HM组为(3.84±0.66) h,明显少于NM组[(6.34±1.78) h;t=2.95,P=0.032],NM组明显少于KF组[(17.64±4.11) h;t=6.86,P=0.001]。NL、NM、NH组的Cmax和AUC0~31 d分别为(321.51±91.86) ng/ml、(719.33±123.70) ng/ml、(1637.23±148.35) ng/ml和(1517.63±386.34) ng·d-1·ml-1、(3479.97±1013.54) ng·d-1·ml-1、(6485.15±1249.98) ng·d-1·ml-1,均与剂量的增加呈线性增加关系[Cmax(r2=0.9981)、AUC(r2=0.9998)]。D1组连续给药约15 d后药物浓度开始维持稳定在1.1 μg/ml左右,28 d 后各组织(脂肪、脾、肺、肾)中的药物浓度分别为(5528.11±106.34) ng/g、(3514.24±45.96) ng/g、(3199.45±155.64) ng/g、(2384.14±55.16) ng/g,均明显高于血浆中药物浓度[(1141.81±50.75) ng/ml;t值分别为-83.24、-77.48、-37.06、-28.11,P值均<0.01]。且各组脂肪组织中药物浓度明显高于其他组织。大鼠经解剖后可见体内脂肪组织均出现黄染,黄染强度依次为D1>NH>NL,肺和脾可见轻微增大。结论 单次给药氯法齐明的药物代谢动力学特征受食物中高脂肪成分及剂量影响较大,多次给药药物浓度在血浆和组织中的分布不均具有独特性,连续给药约15 d后血浆药物浓度维持稳定低值,但组织中药物浓度继续增加。
氯法齐明; 药代动力学; 大鼠,Sprague-Dawley; 评价研究
氯法齐明(clofazimine,Cfz),又名氯苯吩嗪,临床上广泛用于治疗多菌型麻风和麻风性结节性红斑等各类麻风病[1]。1957年Barry等[2]首次报道合成Cfz,因其体外研究显示对结核分枝杆菌(MTB)具有强大的杀菌活性,期望将其用于治疗结核病;但后来在动物体内(豚鼠和恒河猴)研究时未显示出明显的抗MTB活性,加之当时一线抗结核药物刚被研发出来,Cfz在应用于抗结核治疗方面被冷落。随着耐多药结核病(MDR-TB)疫情的暴发,结核病控制形势日益严峻。现有一线抗结核药物多已耐药,二线抗结核药物不良反应较大,而新药研制也困难重重。因此,研究人员开始重新评估Cfz在治疗MDR-TB中的作用。特别是来自孟加拉国的研究者报道了将Cfz 和现有抗耐药结核病药物(5~6种)联合的短程治疗方案(至少9个月)用于MDR-TB患者,取得了87.9%的治愈率,远高于WHO推荐的MDR-TB治疗方案(至少20个月)仅能达到的不足50%的治愈率[3]。该项治疗结果吸引众多研究学者将目光和精力集聚于联合Cfz治疗MDR-TB的短程化疗方案的应用研究中[4-5]。最新WHO耐药结核病治疗指南(2016)[6]首次将Cfz列入了MDR-TB治疗方案中其他二线核心药物,肯定了Cfz对MDR-TB甚至是广泛耐药结核病具有良好的治疗效果,但同时提醒在使用过程中需密切注意其不良反应。本研究旨在通过研究大鼠体内单次或多次给药的药代动力学,获得Cfz血药浓度-时间曲线和药物代谢动力学参数,掌握Cfz在动物体内吸收、分布、代谢、蓄积和排泄的过程和影响因素,观察Cfz在组织中的分布特征,探讨药物作用机制和皮肤着色等不良反应发生的原因,为Cfz在临床应用中的个体化给药剂量调整提供依据。
材料和方法
一、实验材料
1.药物与试剂:Cfz软胶囊(南京立业制药股份有限公司;批号:1212071);Cfz标准品(规格:50 mg,中国食品药品检定研究院;批号:101139-201101);芝麻油(金龙鱼,溶剂);高脂饲料(自制,包括:普通饲料78.8%、胆固醇1%、牛胆盐0.2%、蛋黄粉10%、猪油10%);超纯化水(Milli-Q制备);甲醇(色谱纯,德国Merck公司);甲酸(色谱纯,德国Merck公司)。
2.实验仪器:Agilent1200高效液相色谱仪(美国Agilent科技公司)配备色谱柱Agilent ZORbax SB-C18(3.5 μm, 3.0 mm×100 mm);QTRAP 4500复合三重串联四级杆线性离子阱质谱,配备电喷雾离子源(ESI)及Analyst 1.6数据处理软件(美国AB SCIEX公司);5810R型台式高速冷冻离心机(德国 Eppendorf 公司);5424R型低温冷冻离心机(德国Eppendorf公司);XR205SM-DR型电子分析天平(瑞士Precisa公司);Genius 3型涡旋仪(德国IKA公司);Milli-Q超纯水处理系统(德国IKA公司)。
3.实验动物与饲料:无特定病原体(SPF)级 Sprague-Dawley(SD)雄性大鼠40只,平均体质量(200±20) g,购于广东省医学实验动物中心 [SCXK(粤)2013-0002]。大鼠按随机数字表法分成8组,分为单次给药实验组,包括空白对照组(BL)、空腹实验组(KF)、普通饮食组(NM)、高脂饮食组(HM)、低剂量组(NL)、高剂量组(NH)、中剂量组(即普通饮食组NM);多次给药实验组包括空白对照组(D0)和多次给药组(D1)。每组5只,适应性喂养1周后开始实验,给药前禁食不禁水12 h。所有动物实验符合动物伦理学要求。
二、实验方法
1.给药剂量和途径:按实验设计给药剂量灌胃给予1 ml相应浓度的Cfz油溶液或芝麻油。剂量设计根据临床上成年人(60 kg)一般给药剂量为100 mg/d[7],按体表面积系数换算法,大鼠(0.2 kg)单次给药剂量为2.1 mg/d(10.5 mg·kg-1·d-1),为便于检测将此剂量定为本实验中的低剂量;中剂量组与高剂量组给药剂量分别6.3 mg/d和12.6 mg/d,其他药物试验组给药剂量均设为中剂量6.3 mg/d。空腹实验组给药后再禁食12 h,除高脂实验组以自制高脂饲料喂养,其他组均以普通饲料喂养。多次给药实验组连续给药或溶剂28 d,给药剂量为2.1 mg/d。
2.药物代谢动力学研究:单次给药组给药后采集第0 h、1 h、4 h、8 h、1 d、2 d、4 d、7 d、10 d、14 d、21 d、28 d、31 d的尾静脉血;多次给药组给药后采集第0 h、8 h、1 d、4 d、7 d、10 d、14 d、21 d、28 d的尾静脉血,每次采血0.5 ml并置于肝素抗凝管中,4 ℃下 12 000×g离心 8 min,分离血浆,于-80 ℃保存,待处理。
3.组织分布研究:将全部单次给药组31 d和多次给药组D0、D1组28 d大鼠处死后立即解剖,取脑、心、肝、脾、肺、肾、皮下组织、脂肪(腹部脂肪、肠系膜淋巴结下脂肪、股骨脂肪)、十二指肠、空肠或回肠、肠系膜淋巴结、胰腺、大腿骨共13种组织,用生理盐水洗净组织表面浮血,滤纸吸干。定量称取肝、脾、肺、肾、脂肪等5种组织样品0.5 g(不足0.5 g的组织称量其实际质量),剪碎后放入匀浆管内,加入4倍量的生理盐水于匀浆机上匀浆,取出匀浆液于-80 ℃保存,待处理。
4.生物样品的前处理:(1)血浆样品的处理:取大鼠血浆50 μl,加入100 μl的50%甲醇于1.5 ml离心管中,涡旋2 min,4 ℃下9500×g离心10 min;取上清液50 μl,加入50 μl的100%甲醇,涡旋2 min,4 ℃ 9500×g离心10 min;取60 μl上清液经0.22 μm微孔滤膜过滤,取滤液进行液相色谱-质谱联用法(LC-MS/MS)分析测定药物浓度[8]。(2)组织样品处理:取大鼠组织匀浆液50 μl,加入100 μl的50%甲醇于1.5 ml离心管中,涡旋2 min,4 ℃下9500×g离心10 min;取上清液50 μl,加入50 μl的100%甲醇,涡旋2 min,4 ℃ 9500×g离心10 min;取60 μl上清液经0.22 μm微孔滤膜过滤,取滤液,检测组织中药物浓度方法和血样基本一致。
5. LC-MS/MS法检测药物浓度:使用色谱柱为Agilent ZORBAX SB-C18色谱柱(3.0 mm×100 mm,3.0 μm),柱温:30 ℃。流动相A为0.1%甲酸-甲醇溶液,流动相B为0.1%甲酸-水溶液。梯度洗脱方式为0.00~1.50 min时使用体积百分比为20%A和80% B;1.60~3.00 min之间使用体积百分比为70%A和30% B;3.10~4.00 min之间使用体积百分比为20%A和80% B。流速为700 μl/min。进样量为1 μl。质谱条件采用ESI正离子模式,离子喷射电压为5500 V,离子源温度为550 ℃,去簇电压(DP)为150 V,碰撞能(CE)为50 eV。 采用动态多反应监测模式(MRM)监测定量,定量离子为473.1→431.2质荷比(m/z)。
用甲醇配制Cfz 1 mg/ml的储备液,用甲醇稀制成已知药物浓度的标准溶液,通过在空白血清或空白组织匀浆液中加入不同体积的药物标准溶液,得到含Cfz不同浓度的溶液,分别为500、250、125、62.5、31.25、0 ng/ml,通过LC-MS/MS法检测,以峰面积为纵坐标,药物浓度为横坐标,绘制标准曲线。方法专属性与特异性经检测血浆及组织中的内源性物质均不干扰Cfz的测定,精密度和回收率(n=6)经验证符合方法学的要求,线性范围为0~500 ng/ml。
三、统计学分析
结 果
一、各组血药浓度-时间曲线及药物代谢动力学参数
大鼠单次给予Cfz油溶液后,各实验组均快速吸收达Cmax值。除KF组需17 h外,其他组均在8 h之内达到Cmax值,随后血清药物浓度快速下降至一定值后可维持低浓度达28 d(图1,2)。经DAS 2.0软件按非房室模型统计距法计算得到主要药物代谢动力学参数值,见表1。饮食方式影响实验组(KF、NM、HM):可见Cmax与Tmax值差异均较大,高脂饮食可将空腹Cmax提高约200%以上,同时减少Tmax并增加AUC值约56%(即提高生物利用度)。经统计学分析显示,与NM组Cmax值相比, KF组(t=2.41,P<0.01)和HM组(t=5.77,P<0.01)的差异均有统计学意义;与NM组Tmax值相比,KF组(t=6.86,P=0.001)和HM组(t=2.95,P=0.032)差异也有统计学意义。给药剂量影响实验组(NL、NM、NH):可见单次给药后,随着剂量的增加,药物的Cmax和AUC0~31 d同时增加,并呈线性增加关系[Cmax(r2=0.9981)、AUC(r2=0.9998)]。多次给药组血药浓度逐渐增加,约15 d后浓度维持在1.1 μg/ml左右(图3),这与一般药物多次给药后血药浓度-时间曲线明显不同。
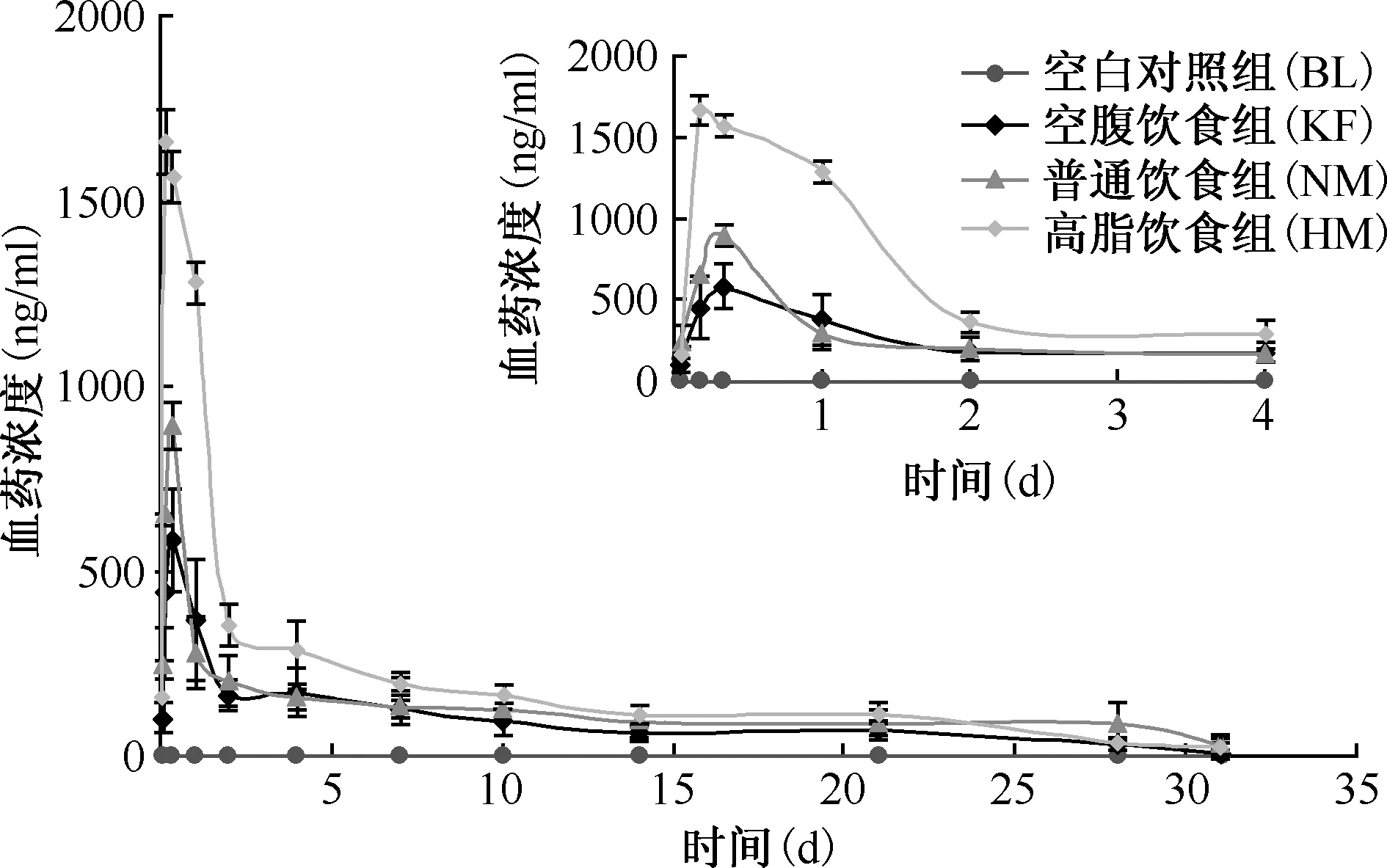
图1 不同饮食方式组大鼠的血药浓度-时间曲线
二、Cfz在体内的分布
与BL组或D0组比较,各实验组大鼠脂肪组织均出现明显着色反应(图4)。D1组连续给药28 d后,脂肪组织(皮下、腹部、肠周围)可见明显橘黄色,NL组单次给药31 d后脂肪组织仅可见轻微淡黄色,NH组单次给药31 d后脂肪组织可见淡黄色。 Cfz在脂肪处的着色与原药本身的橘黄色相近,仅深浅不一,剂量越大,疗程越长,颜色越明显,说明黄染强度与剂量、疗程呈正相关。从局部组织器官解剖图中可见,D1组连续给药后的大鼠肺和脾脏出现明显增大,且肺部有少许深红色物质沉积。各实验组组织药物浓度值见表2所示。单次给药31 d后,各组中仍然能检测到药物浓度,但浓度值均已在最低抑菌浓度(MIC)值(0.25 μg/ml)以下;D1组连续给药28 d后,脂肪(t=-83.24,P<0.01)、脾(t=-77.48,P<0.01)、肺(t=-37.06,P<0.01)、肾(t=-28.11,P<0.01)等组织中的药物浓度均高于血浆中药物浓度(1141.81±50.75) ng/ml,差异有统计学意义;且各组大鼠脂肪中药物浓度明显高于其他组织(NL组:F=50.26,P<0.01;NM组:F=52.88,P<0.01;NH组:F=63.50,P<0.01;多次给药组:F=1639.16,P<0.01)。
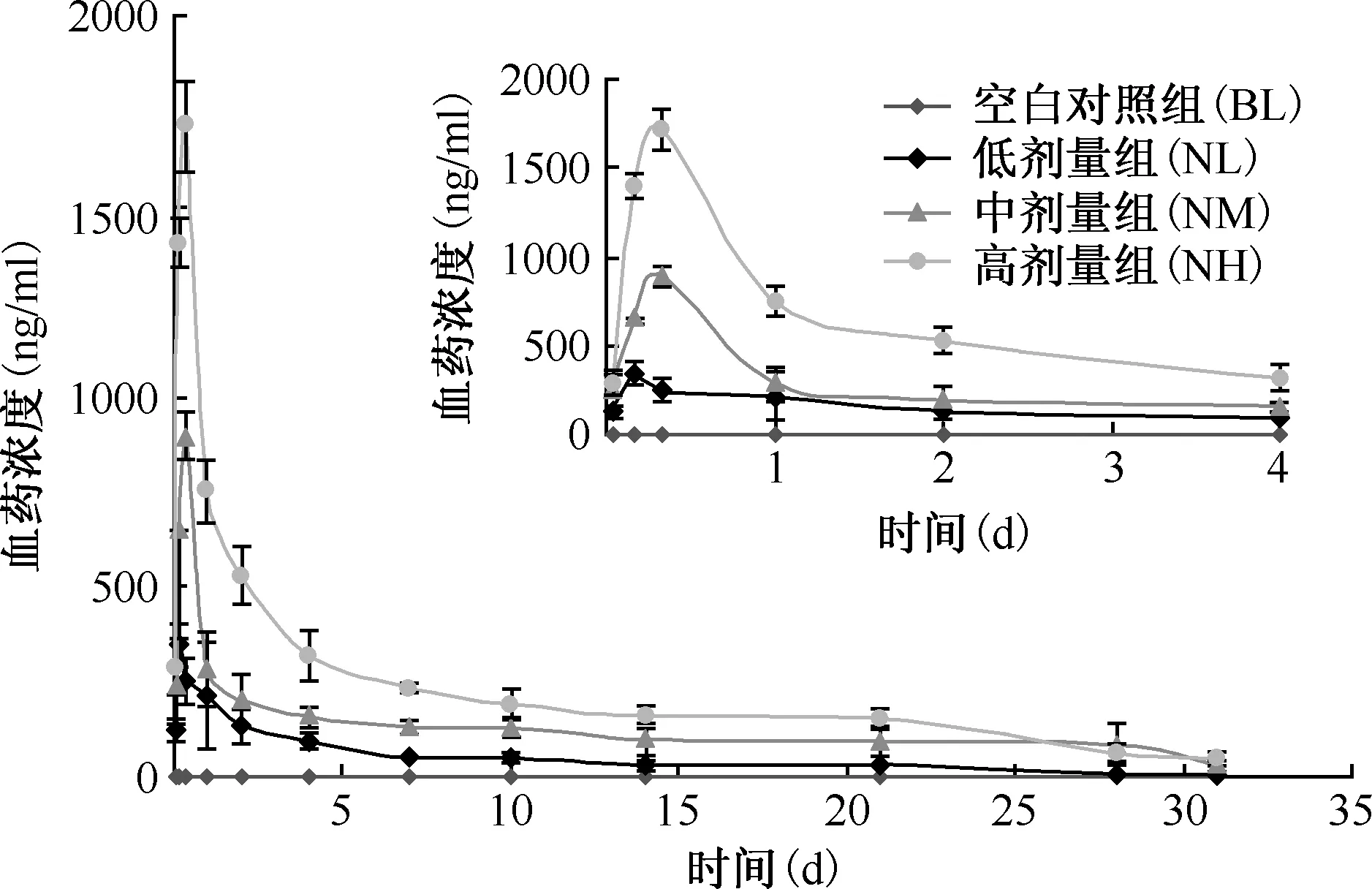
图2 不同给药剂量组大鼠的血药浓度-时间曲线
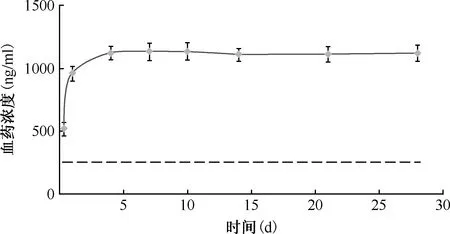
虚线代表Cfz的最低抑菌浓度(MIC)值,为0.25 μg/ml[11]图3 连续多次给药组大鼠的血药浓度-时间曲线
表1 单次给药后各组药物代谢动力学参数
注Cmax:血药峰浓度;t1/2:药物消除半衰期;AUC0~31 d:31 d内药物浓度-时间曲线图下面积;AUC0-inf:DAS2.0软件预测药物浓度-最大时间曲线下面积;MRT:体内平均驻留时间
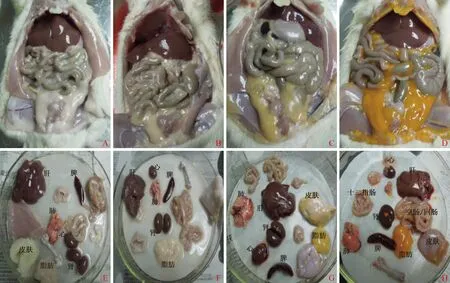
A:空白对照组(BL);B:单次给药(10.5 mg/kg)31 d后(NL组);C:单次给药(63 mg/kg)31 d后(NH组);D:连续多次给药(10.5 mg·kg-1·d-1)28 d后(D1组);图E~H分别为与图A~D相应的解剖组织。与BL组或D0组比较,各实验组大鼠脂肪组织均出现明显着色反应图4 大鼠组织解剖图
表2 不同给药组大鼠各种组织中的药物浓度情况
讨 论
目前,Cfz在抗结核领域的研究多集中于抗MDR-TB临床疗效的观察性研究,对其体内药物代谢动力学过程及药效研究的报道较少,尚无最佳给药剂量指导依据[9]。且患者服用Cfz期间易出现全身皮肤黏膜着色的不良反应,严重影响患者的依从性。我国耐药结核病化学治疗指南(2015)[10]仍将Cfz划分为第五组抗结核药物,指出仅在当Ⅰ~Ⅳ组药物不足以组成有效的耐多药结核病化学治疗方案时才可酌情选用。笔者旨在通过对大鼠体内的初步药物代谢动力学研究,掌握Cfz在体内吸收过程的影响因素及体内分布特征与发生皮肤黄染的原因。本实验研究结果显示,Cfz单次给药后的药物代谢动力学参数与文献报道基本一致[11],多次给药后的血药浓度结果与文献[12]报道一致。大鼠单次给药后,血药浓度在3~8 h之间达到峰值,随后下降至一定低值后会稍微增加再缓慢降低,呈现双指数式释放特征。高脂肪食物可以提高空腹服药的生物利用度达56%,且可增加Cmax并减少Tmax,应该与Cfz较强的亲脂性有关。组织分布以皮毛脂肪组织最为明显,其次为脾、肺、肾等组织。Cfz易蓄积于组织内也应与Cfz的脂溶性密切相关,在经口服吸收入血后随血液向全身各组织器官迁移过程中,由于亲脂性会首先沉着于脂肪组织和单核吞噬细胞系统内,以药物-结晶包合物的药库形式存在[12],随着给药剂量的增加,组织中的药物浓度逐渐增加,原药的棕红色引起脂肪组织黄染;迁移至组织细胞内的药物贮库,将缓慢代谢以被动扩散的方式释放入血被消除,这可能也是导致多次给药后血清药物浓度可以长时间维持稳定浓度状态的原因,也是本文中大鼠经单次给药后呈双指数式释放特征的原因,与药物的二次分布有关。组织中的药物“贮库”的存在,形成了组织中药物浓度明显高于血清中药物浓度的现象。如Srikanth等[13]报道了Cfz在大鼠体内经单次口服20 mg/kg后在骨髓细胞中5 d的暴露量约为血清中的1.5倍;Tyagi等[5]在小鼠结核模型中采取含Cfz的2H-R-Z-C/2H-R-C方案治疗2、3、4个月后,血清中药物浓度分别为1.49、1.26、1.22 μg/ml(与本研究结果基本一致),脾脏中的药物浓度分别为9.04、34.23、416.76 μg/g,停药6个月后,血清中已检测不到Cfz,而脾脏中仍能检测到高于MIC的Cfz,说明脾脏是除脂肪组织外药物易蓄积的第二大组织,与本实验中多次给药组出现脾增大现象一致。药物消除排泄缓慢,单次给予10.5 mg/kg 药物31 d后脂肪组织中黄色仍未完全消退,连续给药后消除半衰期至少为70 d[14],约1~2年才能消退。
在Cfz的早期杀菌活性(early bactericidal activity,EBA)研究中表明,在给药后的2周内均未表现出明显的杀菌活性,而2周后表现出与剂量无关的强大杀菌活性[14];在含Cfz的联合用药治疗研究中表明,联合Cfz治疗方案需2周后才能显示出高于标准治疗方案中的杀菌活性[11]。Cfz多次给药后血药浓度逐渐增加,达到稳定血药浓度(约1.1 μg/ml)所需的时间也约为2周,而2周后Cfz才会显示出杀菌活性,说明Cfz杀菌活性的“有无现象”可能并不取决于血液中的药物浓度,而取决于组织中药物浓度;当组织中蓄积足够的药物剂量(远高于MIC)后,血液中药物浓度才能达到稳定值。Baik等[12]研究表明,蓄积于肝、脾等组织中的Cfz,能与细胞膜作用进入单核吞噬细胞内由晶体结构状药物包合物形成自噬体药物-膜凝集物,诱发抗炎免疫应答反应从而使细胞自杀式凋亡,产生杀菌活性。Cfz的作用机制与作用靶点目前虽尚不明了,但能说明与Cfz在组织中的蓄积量有着紧密的联系,可能存在一个有效组织药物浓度阈值(远高于MIC值),通过小剂量连续多次给药2周之后也能达到这样一个阈值;达到有效浓度之后,又因组织中的药物消除速度非常缓慢,消除半衰期至少为70 d,提示临床可以通过间歇给药或者提前中止给药的短程治疗方案达到有效的杀菌效果,从而减少用药总剂量。为避免剂量相关的着色不良反应,提高患者依从性,在保证Cfz抗结核疗效的前提下,可尽可能地降低给药剂量。有效浓度阈值和临床给药剂量方案的确定尚需更进一步的药物代谢动力学和药效学实验研究。
[1] Cholo MC, Steel HC, Fourie PB, et al. Clofazimine: current status and future prospects. J Antimicrob Chemother, 2011, 67(2): 290-298.
[2] Barry VC, Belton JG, Conalty ML, et al. A new series of phenazine (rimino-compounds) with high antituberculosis activity. Nature, 1957, 179(4568): 1013-1015.
[3] Van Deun A, Maug AK, Salim MA, et al. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med, 2010, 182(5): 684-692.
[4] 荆玮, 王庆枫, 初乃惠. 氯法齐明治疗耐药结核病的研究进展. 中华结核和呼吸杂志, 2016, 39(5):396-399.
[5] Tyagi S, Ammerman NC, Li SY, et al. Clofazimine shortens the duration of the first-line treatment regimen for experimental chemotherapy of tuberculosis. Proc Natl Acad Sci U S A, 2015, 112(3): 869-874.
[6] World Health Organization.WHO treatment guidelines for drug-resistant tuberculosis, 2016 update. Geneva: World Health Organization, 2016.
[7] Schaad-Lanyi Z, Dieterle W, Dubois JP, et al. Pharmacokinetics of clofazimine in healthy volunteers. Int J Lepr Other Mycobact Dis, 1987, 55(1): 9-15.
[8] Kim HJ, Seo KA, Kim HM, et al. Simple and accurate quantitative analysis of 20 anti-tuberculosis drugs in human plasma using liquid chromatography-electrospray ionization-tandem mass spectrometry. J Pharm Biomed Anal, 2015, 102: 9-16.
[9] Gopal M, Padayatchi N, Metcalfe JZ, et al. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int J Tuberc Lung Dis, 2013, 17(8): 1001-1007.
[10] 中国防痨协会. 耐药结核病化学治疗指南(2015). 中国防痨杂志, 2015, 37(5): 421-469.
[11] Swanson RV, Adamson J, Moodley C, et al. Pharmacokine-tics and pharmacodynamics of clofazimine in a mouse model of tuberculosis. Antimicrob Agents Chemother, 2015, 59(6): 3042-3051.
[12] Baik J, Stringer KA, Mane G, et al. Multiscale distribution and bioaccumulation analysis of clofazimine reveals a massive immune system-mediated xenobiotic sequestration response. Antimicrob Agents Chemother, 2013, 57(3): 1218-1230.
[13] Srikanth CH, Joshi P, Bikkasani AK, et al. Bone distribution study of anti leprotic drug clofazimine in rat bone marrow cells by a sensitive reverse phase liquid chromatography method. J Chromatogr B Analyt Technol Biomed Life Sci, 2014, 960: 82-86.
[14] 陆宇, 郑梅琴, 王彬,等. 异烟肼对氯法齐明在小鼠体内组织分布和蓄积的影响. 中华结核和呼吸杂志, 2009, 32(9): 694-697.
(本文编辑:李敬文)
Pharmacokinetics and tissue distribution of clofazimine in rats
FUHan,YANGChun-yan,WANGJun,ZHANGXiao-ping,YANGYing-zhou.
ShenzhenCenterforChronicDiseasePrevention,Shenzhen518020,China
YANGYing-zhou,Email:szyyz@china.com
Objective To explore the pharmacokinetics and tissue distribution of clofazimine (Cfz) in rats. Methods Forty Sprague-Dawley male rats at the weight of (200±20) g were randomly divided into 8 groups (n=5). Single dose groups including rats treated with blank control (BL), fasting group (KF), normal diet group (NM), high fat diet group (HM), low dose group (NL), high dose group (NH), middle dose group (normal diet group, NM); multiple dosing group including blank control group (D0) and multiple drug group (D1). Single dose groups were treated with middle dose of 6.3 mg/d, except 2.1 mg/d and 12.6 mg/d for NL and NH, respectively, and the dose of multiple-dose groups was 2.1 mg/d; while BL and D0 groups were given oil soluble liquid. Blood and tissue samples were collected at different times after single or continuous administration. Serum Cfz levels and drug tissue contents were determined by LC-MS/MS methods. Pharmacokinetic parameters (AUC,Cmax,Tmax, andT1/2) were calculated. Results With single-dose,Cmaxof Cfz in NH group was significantly higher than those in NM group ((1348.05±208.46) ng/ml vs. (719.33±123.70) ng/ml;t=5.77,P<0.01) and KF group ((1348.05±208.46) ng/ml vs. (557.73±85.11) ng/ml;t=2.41,P<0.01);Tmaxof Cfz in NH group was significantly less than that in NM group ((3.84±0.66) h vs.(6.34±1.78) h;t=2.95,P=0.032), whileTmaxof Cfz in NM group was significantly less than that in KF group ((6.34±1.78) h vs.(17.64±4.11) h;t=6.86,P=0.001).Cmaxand AUC0-31 dof NL, NM, NH groups were (321.51±91.86) ng/ml, (719.33±123.70) ng/ml, (1637.23±148.35) ng/ml and (1517.63±386.34) ng·d-1·ml-1, (3479.97±1013.54) ng·d-1·ml-1, (6485.15±1249.98) ng·d-1·ml-1, respectively, which were increased with the dose and showed the relationship of linear increase (Cmax(r2=0.9981), AUC(r2=0.9998)). In D1 group, Cfz level in serum was stable at about 1.1 μg/ml after 15 days of continuous administrations; while 28 days later, the Cfz concentration in tissues was greater than in the plasma ((1141.81±50.75) ng/g), including adipose tissue ((5528.11±106.34) ng/g;t=-83.24,P<0.01), spleen ((3514.24±45.96) ng/g;t=-77.48,P<0.01), lungs ((3199.45±155.64) ng/g;t=-37.06,P<0.01) and kidney ((2384.14±55.16) ng/g;t=-28.11,P<0.01), respectively. And the concentraion in adipose tissue was greater than any other tissues. The adipose tissue of the mice was obviously orange in color, and the intensity of coloration were D1>NH>NL. The spleen and lung was seen slightly increased in the size. Conclusion The pharmacokinetic characteristics of Cfz after one single dose are greatly influenced by the high fat content in food and dosage, which was unique for multiple dosing revealing drug concentration in plasma and tissue maldistribution. After continuous adminstration for about 15 days, the plasma drug concentration was kept stable at low value, but drug concentration in tissue was continue to increase causing accumulation within tissues (especially in adipose tissue) lead to coloration.
Clofazimine; Pharmacokinetics; Rats, Sprague-Dawley; Evaluation studies
10.3969/j.issn.1000-6621.2017.08.013
深圳市卫生和计划生育委员会科研项目(201401068)
518020 深圳市慢性病防治中心
杨应周,Email:szyyz@china.com
2016-11-29)
猜你喜欢
实用癌症杂志(2022年12期)2022-12-26
医学理论与实践(2022年21期)2022-11-10
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年9期)2021-08-24
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年6期)2021-07-31
中国现代医药杂志(2020年10期)2020-12-14
中国食用菌(2020年9期)2020-11-11
中国药物应用与监测(2020年2期)2020-05-28
临床检验杂志(电子版)(2020年1期)2020-04-03
