
利用CRISPR/Cas9技术敲除小鼠ES细胞H2-K1基因*
2017-06-10 08:41陈瑞俊李蔚然黄乙涓刘建中李亮平
中山大学学报(自然科学版)(中英文) 2017年2期
陈瑞俊, 李蔚然, 黄乙涓 , 刘建中, 李亮平,2
(1. 中山大学中山医学院生物学教研室,广东 广州 510080;2. 暨南大学附属第一医院临床医学研究院,广东 广州 510632)
利用CRISPR/Cas9技术敲除小鼠ES细胞H2-K1基因*
陈瑞俊1, 李蔚然1, 黄乙涓1, 刘建中1, 李亮平1,2
(1. 中山大学中山医学院生物学教研室,广东 广州 510080;2. 暨南大学附属第一医院临床医学研究院,广东 广州 510632)
利用CRISPR/Cas9和ES细胞技术,在小鼠ES细胞株上敲除小鼠H2-K1基因,为研发MHCⅠ类基因人源化小鼠打下基础。设计了两个单向导RNA(singleguideRNA,sgRNA),分别靶向H2-K1基因的外显子2和外显子3。以pX330质粒为骨架构建表达sgRNA的打靶载体。用电穿孔转染法将构建好的质粒以及pSUPER-puro共同导入小鼠ES细胞中,实现基因敲除。在嘌呤霉素筛选后,利用PCR初步检测靶基因的敲除情况,再通过测序以及流式细胞分析确定敲除H2-K1基因的小鼠ES细胞。结果显示:利用CRISPR/Cas9技术,小鼠ES细胞的H2-K1基因被成功敲除,通过PCR检测到4个克隆为H2-K1单等位基因敲除(19.0%),2个克隆为H2-K1双等位基因敲除(9.5%)。经过测序以及流式细胞分析,2株小鼠ES细胞被确认为H2-K1双等位基因敲除。本研究利用CRISPR/Cas9技术得到H2-K1基因敲除的小鼠ES细胞株, 为MHCⅠ类基因的敲除和置换提供了参考。
H2-K1 基因;CRISPR/Cas9技术;mES细胞;基因敲除
肿瘤免疫治疗是通过生物制剂直接或间接地诱发或者增强机体的抗肿瘤免疫应答以及破坏肿瘤的免疫抑制效应,达到抑制肿瘤生长或者直接杀伤肿瘤细胞的效果[1]。肿瘤免疫治疗的进展之一是特异性T细胞免疫治疗,而T细胞肿瘤免疫法中一个很重要的进展是T细胞受体(Tcellreceptor,TCR)基因治疗的发展。TCR基因治疗的主要目的是将病人原代T细胞改造成为抗肿瘤或抗感染性疾病的T细胞,实现对疾病的治疗[2]。TCR基因治疗的主要瓶颈是如何获得高亲和力的抗肿瘤TCR基因。李亮平等[3]构建了整个人的TCR-HLA(humanleukocyteantigen)基因位点的基因人源化小鼠,并利用该小鼠模型成功地建立起可以分离得到高亲和力治疗性TCR分子的基础系统。TCR-HLA小鼠的构建需要实现HLA基因的人源化。HLA基因为多等位基因,有将近30种常用的HLA等位基因。这些等位基因将被用于构建HLA基因人源化小鼠,为肿瘤免疫治疗的研发提供了极为重要的基础,和更广泛的应用群体。
小鼠的MHC(majorhistocompatibilitycomplex,MHC)基因称为H-2(histocompatibility-2)基因,人的MHC基因称为HLA基因。小鼠H-2Ⅰ类基因包括:H-2K、H-2D和H-2L等,在一些小鼠中,H-2L基因没有功能。H2-K1基因是H-2K基因的一种单体型(haplotype)。作为MHCⅠ类分子,其表达产物H-2Kb能将内源性抗原肽(如肿瘤抗原肽)提呈到细胞表面,供T细胞的TCR识别[4-5]。1993年,科学家证实,在HLA转基因小鼠体内,如果小鼠自身H-2Ⅰ类分子所限制的免疫应答被抑制,HLAⅠ类分子所限制的免疫应答会很活跃而且多样[6]。根据这一结果,科学家开始研发不表达H-2Ⅰ类分子而只表达HLAⅠ类分子的基因工程小鼠。一种策略是:选取H-2L基因没有功能的小鼠,通过基因敲除分别得到H2-K、H2-D单基因敲除的小鼠;然后让这些小鼠交配得到H2-K、H2-D双基因敲除(即H-2Ⅰ类基因被敲除)的小鼠。H-2Ⅰ类基因被敲除的小鼠和HLAⅠ类转基因小鼠交配,最后得到H-2Ⅰ类基因敲除、HLAⅠ类基因人源化小鼠[7]。实验证明,与H-2Ⅰ类基因没有被敲除的HLAⅠ类转基因小鼠相比,这些小鼠体内HLAⅠ类分子限制的免疫应答被增强[8-10]。这些H-2Ⅰ类分子缺陷的HLAⅠ类基因人源化小鼠在肿瘤免疫治疗、病毒疫苗、基因治疗等领域中有重要的应用[7,11,12]。
CRISPR/Cas9技术来源于细菌和古细菌中的一种干扰破坏入侵的外源性遗传物质的适应性免疫系统,即CRISPR(Clusteredregularlyinterspacedshortpalindromicrepeats)/Cas(CRISPR-associatedproteins)9系统。该系统的主要工作组分包括:Cas9蛋白,crRNA(CRISPRRNA)以及tracrRNA(trans-activatingcrRNA)。在破坏外源性遗传物质时,tracrRNA与crRNA结合形成二聚体并结合在靶序列上,和Cas9蛋白一起介导对靶序列的切割[13]。而CRISPR/Cas9技术对该系统进行了优化,将crRNA和tracrRNA融合成一个sgRNA,由sgRNA与Cas9蛋白介导DNA的靶向切割[14]。无论是CRISPR/Cas9系统还是改造后的CRISPR/Cas9技术,其作用的靶序列附近需要有几个保守的碱基被称为protospacer-adjacentmotif(PAM),PAM通常是:5′-NGG-3′(N为任意一个碱基)[15-16]。现在,CRISPR/Cas9技术已经被用于对细胞以及实验动物的基因组DNA进行基因编辑[14,17-18]。
本研究利用CRISPR/Cas9系统构建了稳定敲除H2-K1基因的小鼠ES细胞(以下简称mES细胞)株,可通过此细胞株得到H2-K1基因敲除的小鼠,为进一步研发有重大应用价值的免疫基因人源化小鼠提供有效的工具。
1 材料与方法
1.1 细胞株、质粒和主要试剂
129小鼠品系胚胎干细胞 (mouseembryonicstem,mES)、怀孕13.5d的ICR小鼠品系胚胎成纤维细胞(mouseembryofibroblast,MEF),购自赛业(广州)生物科技有限公司。pSuperpuro质粒为本实验室保存。pX330质粒,购自Addgene。
大肠杆菌DH5α感受态细胞、氨苄青霉素,购自广州鼎国生物技术有限公司。1kbDNALadder,购自上海生工生物工程技术服务有限公司。琼脂糖凝胶DNA回收试剂盒、pGH平末端PCR产物克隆试剂盒和质粒小量制备试剂盒,购自上海捷瑞生物工程有限公司。FastDigestBbsⅠ限制性内切酶,购自Fermentas。去内毒素质粒中量制备试剂盒,购自Promega公司。基因组DNA提取试剂盒(DNeasyBlood&TissueKit),购自Qiagen公司。QuickTMLigationKit、T4PolynucleotideKinase(T4PNK),购自NewEnglandBiolabs公司。电转试剂盒,AmaxaTMP3PrimaryCell4D-NucleofectorTMXKitL,购自Lonza公司。PrimerStarMax(Takara公司),购自广州瑞真生物公司。寡核苷酸单链合成、PCR引物合成和质粒测序服务,购自Invitrogen公司。流式抗体:FITCMouseAnti-MouseH-2Kb(Isotype:MouseIgG2a,κ)、FITCMouseAnti-MouseH-2Db(Isotype:MouseIgG2b,κ)、FITCMouseIgG2a,κIsotypeControl以及FITCMouseIgG2b,κIsotypeControl,购自BDBioscience公司。胎牛血清、青链霉素、嘌呤霉素、DMEM、Trypsin-EDTA、谷氨酰胺(100×),β-巯基乙醇,非必需氨基酸(100×),丙酮酸钠,购自LifeTechnologies。白血病抑制因子(Leukemiainhibitoryfactor,LIF),购自Millipore。二甲基亚砜(Dimethylsulfoxide,DMSO)、明胶、丝裂霉素C,购自Sigma公司。
1.2sgRNA靶点的选择及其序列的合成
利用哈佛大学张峰实验室(ZhangFengLab,MIT)提供的网络工具CRISPERDESIGN(http://crispr.mit.edu)设计H2-K1基因的向导RNA(sgRNA)靶点。根据H2-K1基因的结构,分别在其外显子2和外显子3上各设计一个sgRNA靶点。根据质粒pX330中BbsⅠ酶切位点的结构,在正义链寡核苷酸(oligo)序列的5′端添加“CACC”,并在反义链寡核苷酸序列的5′端添加“AAAC”, 这与BbsⅠ酶切后形成的黏性末端互补(图1:A)。为增强U6启动子的转录,在其起始转录位点需要添加碱基G[19]。如果正义链寡核苷酸序列的“CACC”后面的碱基不是“G”,则需要添加碱基“G”,并在反义链寡核苷酸序列的3′端添加碱基“C”。
1.3 质粒构建
首先,用BbsⅠ内切酶切割质粒pX330(1~2 μg), 电泳后,按照琼脂糖凝胶DNA回收试剂盒说明书进行切胶回收。将设计的单链oligo退火形成双链,退火体系:1 μL,oligo 1(100 μmol/L);1 μL,oligo 2(100 μmol/L);1 μL,10×T4 Ligation Buffer (NEB);6.5 μL,ddH2O;0.5 μL,T4 PNK (NEB);退火程序:37 ℃,30 min;95 ℃,5 min;-6 ℃/min,降温至25 ℃。将退火形成的双链DNA连接到经BbsⅠ酶切回收后的pX330载体中,连接体系:线性化的pX330,50 ng;退火产物经1∶200稀释,后取1 μL稀释液;2×Quickligation Buffer,5 μL;补水至10 μL;加入1 μL Quick Ligase后,室温连接10 min。将连接产物在42 ℃转化到DH 5α感受态细胞中, 用含有氨苄青霉素的琼脂糖平板进行筛选过夜, 挑取单克隆,提取小制备质粒DNA并通过测序验证插入序列的正确性。
1.4 细胞培养、转染和药筛
MEF细胞培养条件:DMEM+15%(w)胎牛血清+ 1%(w)谷氨酰胺+1 mmol/L丙酮酸钠+ 1%(w)非必需氨基酸+100 U/mL青霉素+100 μg/mL链霉素。37 ℃、φ=5% CO2恒温培养。
用丝裂霉素C处理MEF细胞:往MEF培养基中加入丝裂霉素C,使其质量浓度为10 μg/ mL。待MEF铺板率达到90%以上后,吸除培养基,加入含丝裂霉素C的MEF培养基。37 ℃、φ=5% CO2恒温培养2 h后,吸除培养基,用PBS洗3遍。此时,MEF细胞可用普通MEF培养基培养;也可以消化下来,种在用明胶处理的培养皿中,用于培养mES细胞。
明胶处理培养皿:往100 mm培养皿中加入w=0.1%的无菌的明胶溶液,于37 ℃恒温培养箱中,放置2 h,然后彻底除去明胶溶液。
mES细胞的培养条件:用DMEM+15%(w)胎牛血清+0.1 mmol/Lβ-巯基乙醇+106IU/mL LIF(白血病抑制因子)+1%(w)谷氨酰胺+1 mmol/L丙酮酸钠+1%(w)非必需氨基酸+100 U/mL青霉素+100 μg/mL链霉素的培养基培养mES细胞。在明胶处理好的培养皿中种上MEF细胞,然后换成将mES细胞培养基,将mES细胞种在皿中,37 ℃、φ=5% CO2恒温培养。
mES细胞去除MEF细胞处理:在电转和鉴定打靶的mES时需要去除MEF。用胰酶将mES细胞消化下来。加入培养基终止消化,离心去除胰酶上清后,用mES培养基重悬细胞,转移到明胶处理过的培养皿中,置于37 ℃、φ=5% CO2恒温培养20~30 min,使MEF贴壁生长。然后,轻轻吸出悬液,离心收集悬浮的mES细胞。如果MEF细胞没有除尽,可在mES克隆长出来后,可以重复上述步骤去除MEF细胞,直到可进行下一步实验。
mES的CRISPR/Cas9基因打靶:培养129-mES细胞,使其保持未分化状态。未分化mES细胞团浓密,呈椭圆形或圆形且边界清晰明显;在还没有出现细胞团融合时,用胰酶将ES细胞消化下来。经过去除MEF细胞的处理后,按照“AmaxaTM4D-NucleofectorTMProtocol”中小鼠ES电转化法的使用说明进行电转,电转程序选用4D-NucleofectorTMSystem中已设定的小鼠ES细胞电转程序。收集4×106个细胞,用电转缓冲液重悬;并加入7.5 μg的pX330-H2K1-Exon2,7.5 μg的pX330-H2K1-Exon3,5 μg的pSuper-puro,1 μg的pmaxGFP。电转36 h后,给细胞换上含1 μg/mL嘌呤霉素的mES细胞培养基,进行药筛,在药筛18 h时,补充3×105个经丝裂霉素C处理的MEF细胞。药筛36 h后,给细胞换上普通的mES细胞培养基,并再次补充5×105个经丝裂霉素C处理的MEF细胞,继续培养。
1.5 mES细胞克隆的扩增和基因组DNA的提取
mES细胞集落形成后,挑取单克隆,进行单独培养以及基因型鉴定。每个mES细胞单克隆在扩增后都分成两份,一份用于冻存,一份用于扩增细胞。扩增的ES细胞,去MEF细胞后提基因组DNA进行鉴定。提取基因组DNA时,先消化收集细胞(每个样品约收集6×105个细胞),并用PBS洗两次,按照基因组DNA提取试剂盒说明书的操作步骤提取基因组DNA。
1.6 mES细胞基因打靶的鉴定
用PCR法扩增打靶区域的DNA序列,根据靶点的位置,设计基因敲除鉴定引物:H2-K1-(2,3)-F:CGGATCCGGTGGCGCGATCACCAAGAACCAATC; H2-K1-(2,3)-R:GGAATTCCTGACACATTCAGCAGGACAGGAGTC。
PCR条件如下: Primer Star Max 10 μL, 基因组DNA 1 μL(60 ng), Primers(F+R)(2 μmol/L) 2 μL, 补水至20 μL。程序如下: 第1个循环,95 ℃,变性3 min。95 ℃,变性30 s;57 ℃,退火45 s;72 ℃,延伸1 min 45 s,共35个循环。最后72 ℃延伸5 min, 4 ℃保存。取反应后产物5 μL进行琼脂糖凝胶电泳检测。
对发生了基因打靶的PCR产物进行分子克隆和测序:用基因组DNA做PCR(50 μL×8),对需要检查的条带进行胶回收。用pGH平末端PCR产物克隆试剂盒将PCR目的条带连接入载体,连接反应体系:pGH Blunt end Vector (25 ng/μL),2 μL;插入片段(50 ng/μL),1.5 μL(分子数vector:insert大约为1∶5);2×quick ligation solution,5 μL;ddH2O,1.5 μL;16 ℃连接过夜。将连接产物转化DH5α感受态细胞,接种在含有氨苄青霉素的琼脂糖平板上。37 ℃培养过夜,长出菌落后,挑取单克隆;摇菌,提质粒后送样测序。
1.7 基因打靶细胞表型鉴定
mES细胞分化培养:对确定有H2-K1基因敲除的ES细胞克隆进行分化培养。首先复苏冻存的拷贝,去除mES细胞中的MEF细胞,然后将mES细胞接种在没有经明胶处理的培养皿中,37 ℃、φ=5%CO2恒温培养。用mES细胞培养基培养几天以扩增细胞数量,然后,用mES细胞分化培养基进行分化培养,分化的细胞多数呈纤维状。
细胞表面H-2Kb分子表达的检测:在大部分mES细胞分化后,消化收集细胞,调整细胞浓度为106~107个/ mL,用小鼠MHC分子单克隆抗体(anti-mouse H-2Kb)检测H-2Kb分子的表达。取流式抗体(每种流式抗体各2 μL)加入100 μL的待测细胞悬液中,4 ℃,避光反应30 min。反应完毕后,离心收集细胞,用PBS洗细胞后,加入100 μL PBS重悬细胞,然后上样检测以及分析结果。
2 结 果
2.1 CRSPR/Cas9基因打靶载体的设计与构建
利用哈佛大学张峰实验室提供的网络工具,在H2-K1基因的外显子2和外显子3上各设计了一个sgRNA,并命名为H2-K1-Exon2-sgRNA和H2-K1-Exon3-sgRNA(图1:B)。两个靶位点之间的序列长度为0.56kb。
根据pX330质粒中BbsⅠ酶切位点的结构以及U6启动子的特性,设计了需要合成的寡核苷酸链(表1)。通过退火使寡核苷酸单链形成双链, 然后将退火产物克隆于pX330质粒的BbsⅠ酶切位点上, 得到pX330-H2-K1-Exon2和pX330-H2-K1-Exon3载体;测序后,扩增正确的质粒,用于下一步实验。
2.2 小鼠ES细胞H2-K1基因打靶
消化mES细胞并经过去MEF细胞的处理后,用AmaxaTMP3PrimaryCell4D-NucleofectorTMXKitL电转试剂盒,将pX330-H2K1-Exon2,pX330-H2K1-Exon3,pSuper-puro,pmaxGFP共4个载体共转染mES细胞。电转36h后,在荧光显微镜下可观察到电转后的细胞表达绿色荧光,表示电转染成功,用含1μg/mL嘌呤霉素的mES细胞培养基进行药筛。
警务指挥系统可以很好地处理在交通中产生的应急情况,得到出现事故路线的交通情况和车辆状态,从而将信息及时地发布给相关人员,避免交通出现进一步拥堵。公共交通服务系统可以为出行者进行线路的规划,确定合适的出行方式,从而确保其顺利出行。
筛选后,换普通的mES细胞培养基继续培养mES细胞,5d后挑取mES细胞单克隆并培养扩增。mES细胞扩增后,取一部分用于去MEF细胞、提取基因组DNA并做PCR鉴定H2-K1基因是否被敲除。以野生型的mES细胞(wildtypemEScell)为对照组,标记为:WT。对于野生型DNA、仅发生小片段缺失或插入的DNA,其PCR条带为1.43kb;对于在两靶点间发生整个片段敲除的DNA,其PCR条带为0.87kb。本实验通过PCR鉴定了21个克隆(图2),其中,#3、#5、#9、#20,共4个克隆都在一个等位基因上发生两靶点间的片段敲除(19.0%);#12、#17,共2个克隆在双等位基因上都发生两靶点间的片段敲除(9.5%);总共打靶效率为28.5%(6/21)。
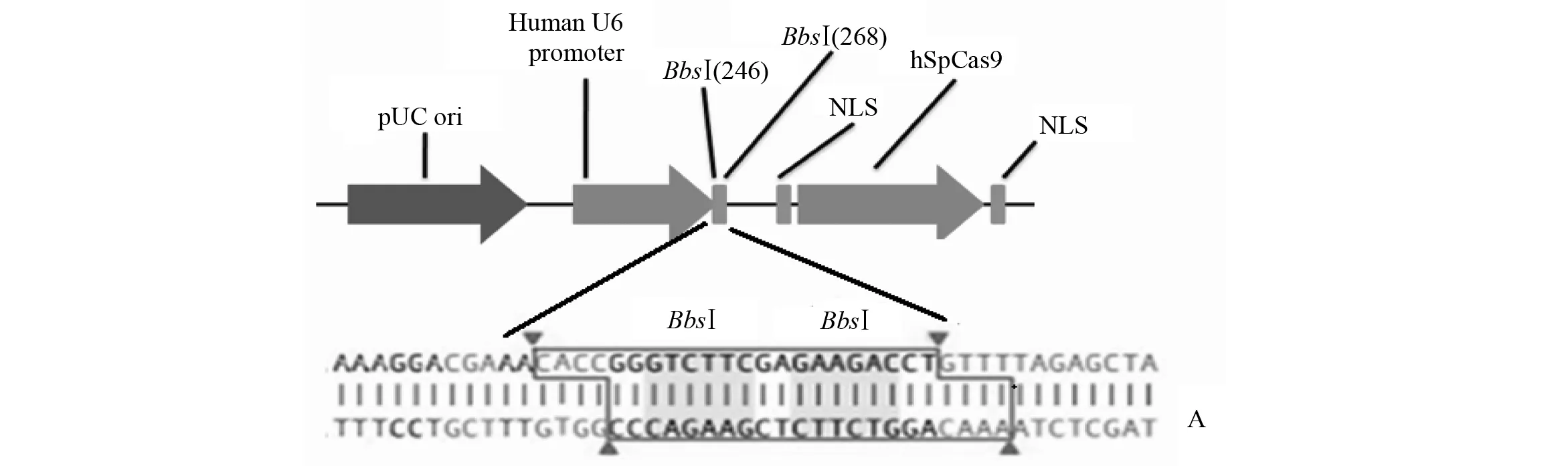
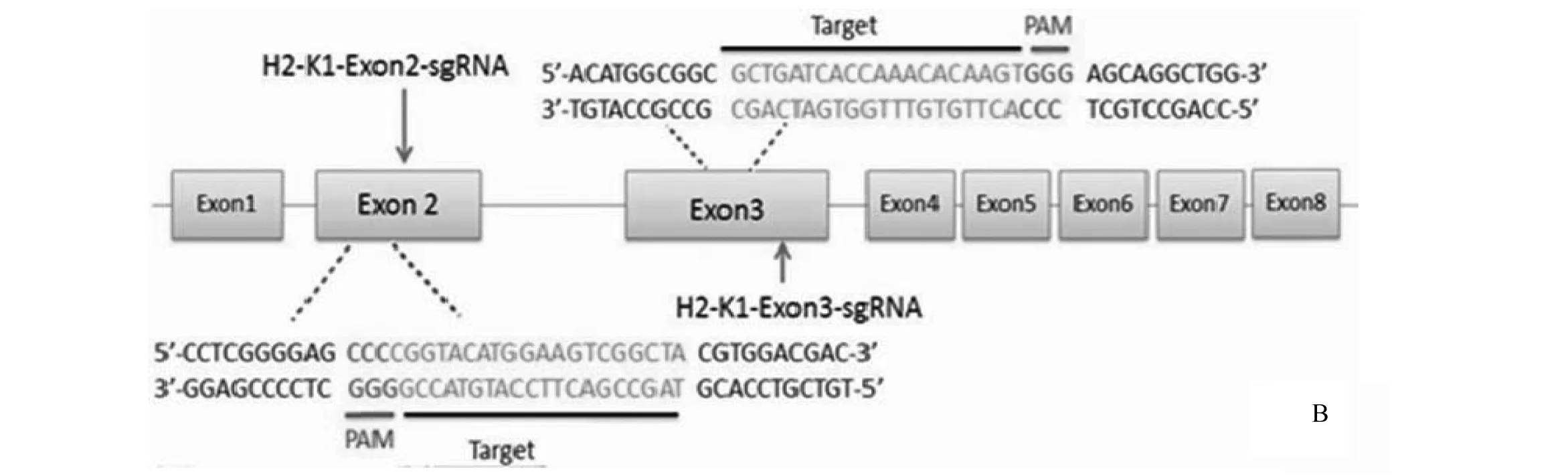
图1 构建靶向H2-K1的pX330质粒Fig.1 Construction of H2-K1-targeting pX330 plamidsA:pX330质粒结构,包含2个BbsⅠ酶切位点以及启动子; B: H2-K1-Exon2-sgRNA以及H2-K1-Exon3-sgRNA的靶位点及其附件序列

sgRNAOligonucleotidesequenceH2-K1-Exon2-sgRNAsenseoligo:5'-CACCGTAGCCGACTTCCATGTACCG-3'antisenseoligo:5'-AAACCGGTACATGGAAGTCGGCTAC-3'H2-K1-Exon3-sgRNAsenseoligo:5'-CACCGCTGATCACCAAACACAAGT-3'antisenseoligo:5'-AAACACTTGTGTTTGGTGATCAGC-3'
2.3mES细胞克隆基因型鉴定
本次研究对#12和#17克隆进行了提取基因组DNA、PCR、连接载体以及测序鉴定。分析测序结果证实, 这两株克隆的靶位点间的片段被敲除。假设特异性切割的位置是在靶位点5′往3′方向的第17和第18个碱基之间(即PAM序列前的第3和第4个碱基之间)[14]。其中,#17克隆有3个靶位点在假设位置上发生了精确的切割(Clone17-Allele-1的H2-K1-Exon2-sgRNA靶点,Clone17-Allele-2的H2-K1-Exon2-sgRNA靶点和H2-K1-Exon3-sgRNA靶点),#12和#17克隆其他5 个靶位点上的靶向切割都发生了小片段的缺失或者插入(图3)。
2.4H2-K1基因表达的鉴定
MHC分子在ES细胞上只有少量的表达,为了便于分析,本次研究对#12、#17mES细胞克隆以及野生型mES细胞克隆进行分化培养,前4~5d用mES细胞培养基培养,ES细胞克隆长出来后,用mES细胞分化培养基培养(图4)。培养25~30d后,大部分的mES细胞发生分化,收集细胞,进行流式细胞术检测(图4)。
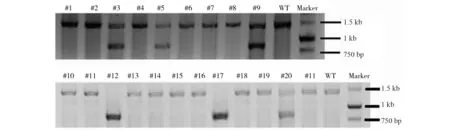
图2 PCR鉴定基因片段敲除的mES细胞克隆Fig.2 The identification of the mES cell clones with gene deletion
3 讨 论
H2-K1基因是小鼠MHCⅠ类基因的一种,其表达产物H-2Kb参与了病毒抗原、肿瘤抗原等内源性抗原的加工和提呈。MHCⅠ类基因人源化小鼠在肿瘤免疫治疗、病毒疫苗、基因治疗等领域中有重要的应用[7,11-12]。CRISPR/Cas9技术是一项新的基因组编辑技术,和锌指核酸酶(zincfingerendonuclease,ZFN)、转录激活因子样效应物核酸酶(transcriptionactivator-likeeffectornuclease,TALEN)一样,可以用于编辑细胞和实验动物的基因组DNA[20-21]。本实验应用CRISPR/Cas9技术,以pX330质粒为骨架,快速地构建了用于敲除H2-K1基因的质粒载体,并且通过电击穿孔转染的方法将载体转染到mES细胞中。在mES细胞单克隆挑取以及细胞扩增后,经PCR、DNA测序以及流式细胞分析证实,本研究得到了两株H2-K1双等位基因敲除的mES细胞株。
本研究选取了一对靶位点,去除的是一段基因。如果只设计一个靶位点,可能会产生小片段碱基的缺失或者插入,但是少数几个碱基的变化不一定能够使基因沉默。而且小片段的突变很难通过PCR的方法来检测,需要通过T7核酸内切酶Ⅰ或者Suveryor试验等方法进行检测,过程比较复杂。与之相比,本次实验去除的基因片段包含了H2-K1基因的Exon2和Exon3的序列,破坏了其对应的分子的α1和α2结构,能够更有效地敲除H2-K1基因。通过普通的PCR方法即可鉴定DNA片段是否被敲除。除此之外,在提供带同源臂的DNA模板的条件下,Cas9蛋白对DNA进行切割产生DNA双链断裂(double-strandbreak,DSB)后,可通过以同源指导修复(homologydirectedrepair,HDR)为基础的DNA修方式,实现对DNA的修饰[17-18]。本实验成功地敲除H2-K1基因两个靶点间的基因片段,证实所设计的两个靶点可以作为CRISPR/Cas9系统的靶点。靶点所在的Exon2和Exon3对应H2-Kb分子的α1和α2结构,正是MHCⅠ类分子与抗原肽结合的部位,这是不少科学家在人源化小鼠的MHC基因时修饰的基因区间[7]。因此,本次实验所设计的两个靶位点为H2-K1基因的修饰提供两个可靠的靶位点。

图4 mES细胞分化培养养Fig.4 The differentiation of mES cellsA:#12 mES细胞分化前的形态;B: #17 mES细胞分化前的形态;C: 野生型mES细胞分化前的形态;D:#12 mES细胞分化后的形态;E: #17 mES细胞分化后的形态;F: 野生型mES细胞分化后的形态

图5 流式细胞术检测结果Fig.5 The result of flow cytometryA、B:蓝色曲线代表用FITC Mouse Anti-Mouse H-2Kb流式抗体标记分化的#12 mES细胞的检测结果;橙色曲线代表用FITC Mouse Anti-Mouse H-2Kb流式抗体标记分化的#17 mES细胞的检测结果;黑色曲线代表用FITC Mouse IgG2a, κ Isotype流式抗体标记分化的野生型mES细胞的检测结果;红色曲线代表用FITC Mouse Anti-Mouse H-2Kb流式抗体标记分化的野生型 mES细胞的检测结果;C、D:紫色曲线代表用FITC Mouse Anti-Mouse H-2Db流式抗体标记分化的#12 mES细胞的检测结果;绿色曲线代表用FITC Mouse Anti-Mouse H-2Db流式抗体标记分化的#17 mES细胞的检测结果;黑色曲线代表用FITC Mouse IgG2b, κ Isotype流式抗体标记分化的野生型mES细胞的检测结果;红色曲线代表用FITC Mouse Anti-Mouse H-2Db流式抗体标记分化的野生型 mES细胞的检测结果
研发MHCⅠ类基因人源化小鼠的一种策略是先分别得到H2-K、H2-D单基因敲除的小鼠。通过显微注射技术,本研究获得的H2-K1双等位基因敲除的mES细胞可以被注射到小鼠早期胚胎内。通过对小鼠的饲养、繁殖以及鉴定,可以得到H2-K1双等位基因敲除的基因工程小鼠。这为MHCⅠ类基因人源化小鼠的研发打下基础。这与本实验组的研发HLA基因人源化小鼠的目的相符。我们在进行本实验的同时在利用基因置换技术开展HLA转基因小鼠的研究,计划将H2基因敲除小鼠与HLA转基因小鼠交配,得到新一代HLA基因人源化小鼠。这一系列HLA基因人源化小鼠模型将为肿瘤免疫治疗提供研究平台。
综上所述,本研究联合使用CRISPR/Cas9技术、电穿孔转染法等技术得到两株H2-K1双等位基因敲除的mES细胞,同时也证实所设计的两个靶位点的有效性。这些获得的mES细胞株、所设计的靶位点以及打靶载体都为MHCⅠ类基因人源化小鼠的研发以及病毒疫苗、基因治疗等研究提供很好的工具。
[1]DOUGANM,DRANOFFG.Immunetherapyforcancer[J].AnnuRevImmunol,2009,27:83-117.
[2]KIEBACKE,UCKERTW.EnhancedTcellreceptorgenetherapyforcancer[J].ExpertOpinBiolTher,2010,10(5):749-762.
[3]LILiangping,LAMPERTJC,CHENXiaojing,etal.TransgenicmicewithadiversehumanTcellantigenreceptorrepertoire[J].NatMed,2010,16(9):1029-1034.
[4]STEVANOVICS,SCHILDH.QuantitativeaspectsofTcellactivation-peptidegenerationandeditingbyMHCclassImolecule[J].SeminarsinImmunology,1999,11(6):375-384.
[5]GIL-TORREGROSABC,CASTANOAR,LOPEZD,etal.GenerationofMHCclassIpeptideantigensbyproteinprocessinginthesecretoryroutebyfurin[J].Traffic,2000,1(8):641-651.
[6]BARRAC,GOURNIERH,GARCIAZ,etal.AbrogationofH-2-restrictedCTLresponsesandefficientrecognitionofHLA-A3moleculesinDBA/2HLA/A24respondermice[J].JImmunol,1993,150:3681-3689.
[7]PASCOLOS.HLAclassItransgenicmice:development,utilisationandimprovement[J].ExpertOpiniononBiologicalTherapy,2005,5(7):919-938.
[8]DAFTRIANP,ALIS,SHARANR,etal.ImmunizationwithTh-CTLfusionpeptideandcytosine-phosphate-guanineDNAintransgenicHLA_A2miceinducesrecognitionofHIV-infectedTcellsandclearsvacciniaviruschallenge[J].JImmunol,2003,171:4028-4039.
[9]PETERK,MENY,PANTALEOG,etal.InductionofacytotoxicT-cellresponsetoHIV-1proteinswithshortsyntheticpeptidesandhumancompatibleadjuvants[J].Vaccine,2001,19:4121-4129.
[10]FIRATH,COCHETM,ROHRLICHPS,etal.ComparativeanalysisoftheCD8(+)TcellrepertoiresofH-2classIwild-type/HLA-A2.1andH-2classⅠknockout/HLA-A2.1transgenicmice[J].IntImmunol,2002,14:925-934.
[11]SEYEDN,TAHERIT,VAUCHYC,etal.ImmunogenicityevaluationofarationallydesignedpolytopeconstructencodingHLA-A*0201restrictedepitopesderivedfromLeishmaniamajorrelatedproteinsinHLA-A2/DR1transgenicmice:Stepstowardpolytopevaccine[J].PLoSOne,2014,9(10):e108848.
[12]BOUCHERMAR,KRIDANE-MILEDIH,BOUZIATR,etal.HLA-A*01:03,HLA-A*24:02,HLA-B*08:01,HLA-B*27:05,HLA-B*35:01,HLA-B*44:02,andHLA-C*07:01MonochainTransgenic/H-2classInullmice:NovelversatilepreclinicalmodelsofhumanTCellresponses[J].JournalofImmunology,2013,191(2):583-593.
[13]JIANGW,MARRAFFINILA.CRISPR-Cas:Newtoolsforgeneticmanipulationsfrombacterialimmunitysystems[J].AnnualReviewofMicrobiology,2015,69:209-228.
[14]CANVERMC,BAUERDE,DASSA,etal.Characterizationofgenomicdeletionefficiencymediatedbyclusteredregularlyinterspacedpalindromicrepeats(CRISPR)/Cas9nucleasesysteminmammaliancells[J].JournalofBiologicalChemistry,2014,289(31):21312-21324.
[15]CENCICR,MIURAH,MALINAA,etal.Protospaceradjacentmotif(PAM)-distalsequencesengageCRISPRCas9DNAtargetcleavage[J].PLoSOne,2014,9(10):e109213.
[16]JINEKM,CHYLINSKIK,FONFARAI,etal.Aprogrammabledual-RNA-guidedDNAendonucleaseinadaptivebacterialimmunity[J].Science,2012,337(6096):816-821.
[17]MAYuanwu,MAJing,ZHANGXu,etal.GenerationofeGFPandCreknockinratsbyCRISPR/Cas9[J].FebsJournal,2014,281(17):3779-3790.
[18]YANGHui,WANGHaoyi,SHIVALILACS,etal.One-stepgenerationofmicecarryingreporterandconditionalallelesbyCRISPR/Cas-mediatedgenomeengineering[J].Cell,2013,154(6):1370-1379.
[19]MALIP,YANGLuhan,ESVELTKM,etal.RNA-guidedhumangenomeengineeringviaCas9[J].Science,2013,339(6121):823-826.
[20] 聂宇, 乔艳乐, 陈瑶生, 等. 供体同源臂长度对ZFN介导的同源重组效率的影响 [J]. 中山大学学报(自然科学版),2016,55(4):100-107.NIEYu,QIAOYanle,CHENYaosheng,etal.TheeffectofthelengthofdonorhomologousarmontheefficiencyofZFN-inducedhomologousrecombination[J].ActaScientiarumNaturaliumUniversitatisSunyatseni,2016,55(4):100-107.
[21] 倪培凌, 刘畅, 陈凌懿.DNA剪刀—TALEN和CRISPR/Cas[J]. 中国细胞生物学学报,2014,36(1):5-11.NIPeiling,LIUChang,CHENLingyi.DNAscissors—TALENandCRISPR/Cas[J].ChineseJournalofCellBiology,2014,36(1):5-11.
TheH2-K1geneknockoutofmouseEScells
throughCRISPR/Cas9technology
CHENRuijun1,LIWeiran1,HUANGYijun1,LIUJianzhong1,LILiangping1,2
(1.Department of Biology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China; 2.Central Laboratory, The First Affiliated Hospital, Jinan University, Guangzhou 510632, China)
The CRISPR/Cas9 technology was used to achieve theH2-K1geneknockoutofmouseEScells,whichwillbeusefulforgenerationofMHCclassⅠgenehumanizedmice.Inthisstudy,twosgRNAsweredesigned,whicharetargetingtotheExon2andExon3ofH2-K1,respectively.TheplasmidsexpressingthesgRNAswereconstructedusingpX330asthematrixplasmid.InordertoknockoutH2-K1,theconstructedplasmidsandpSUPER-purowerecotransfectedintothemEScellsthroughelectroporation.Afterscreeningbypuromycin,theresultofgenetargetingwasdeterminedbyPCR,andH2-K1knockoutmouseEScellswerefurtherconfirmedthroughsequencingandflowcytometry.WefoundthatH2-K1inthemouseEScellwasknockedoutusingCRISPR/Cas9technology.ThroughPCR, 4clonesweredeterminedasonealleleknockout(19.0%),2clonesweredeterminedastwoalleleknockout(9.5%). 2cloneswerefurtherconfirmedastwoalleleknockoutclonesbysequencingandflowcytometry.ThegeneratedH2-K1knockoutmouseEScellswouldprovideareferencefortheknockoutandreplacementofMHCclassⅠgene.
H2-K1gene;CRISPR/Cas9technology;mEScell;geneknockout
2016-09-05 基金项目:国家自然科学基金(31270920;81472824);广东省科技计划项目(2013B060300004)
陈瑞俊(1989年生),男;研究方向:肿瘤免疫学;E-mail:347770864@qq.com
李亮平(1961年生),男;研究方向:肿瘤免疫学;E-mail:lilping2@mail.sysu.edu.cn,liangping_li@jnu.edu.cn
10.13471/j.cnki.acta.snus.2017.02.002
R
A
0529-6579(2017)02-0005-07
猜你喜欢
环球时报(2022-09-20)2022-09-20
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
世界科学技术-中医药现代化(2022年3期)2022-08-22
成都医学院学报(2022年4期)2022-08-19
江西农业学报(2021年4期)2021-04-20
今日农业(2020年24期)2020-12-15
商品与质量(2020年9期)2020-11-26
三农资讯半月报(2020年11期)2020-06-21
食品与生物技术学报(2020年2期)2020-01-05
祝您健康(2018年12期)2018-11-27
