
集成改进KNN算法预测蛋白质亚细胞定位
2017-05-06 07:09薛卫王雄飞赵南杨荣丽洪晓宇
生物工程学报 2017年4期
薛卫,王雄飞,赵南,杨荣丽,洪晓宇
集成改进KNN算法预测蛋白质亚细胞定位
薛卫,王雄飞,赵南,杨荣丽,洪晓宇
南京农业大学信息科学技术学院,江苏南京 210095
基于Adaboost算法对多个相似性比对K最近邻 (K-nearest neighbor,KNN) 分类器集成实现蛋白质的亚细胞定位预测。相似性比对KNN算法分别以氨基酸组成、二肽、伪氨基酸组成为蛋白序列特征,在KNN的决策阶段使用Blast比对决定蛋白质的亚细胞定位。在Jackknife检验下,Adaboost集成分类算法提取3种蛋白序列特征,3种特征在数据集CH317和Gram1253的最高预测成功率分别为92.4%和93.1%。结果表明Adaboost集成改进KNN分类预测方法是一种有效的蛋白质亚细胞定位预测方法。
亚细胞区间,蛋白序列特征,K-nearest neighbor,basic local alignment search tool,Adaboost
蛋白质的功能与其所属的亚细胞定位有着紧密的联系,不同蛋白质只有处于特定的亚细胞定位才能发挥其功能,保障生命活动的正常进行,因此对蛋白序列的亚细胞定位预测研究有着重要意义[1]。
利用机器学习实现蛋白质亚细胞定位预测是目前获取定位信息的主要方法,并取得了一系列进展[2-6]。Zhou等构建凋亡蛋白数据集,在氨基酸组成特征下,利用协变判别函数实现区间预测[7]。Huang等用支持向量机对氨基酸组成特征实现了对凋亡蛋白的预测[8]。Bulashevska等用贝叶斯分类器同样得到较好的分类预测效果[9]。Chen等在使用凋亡蛋白数据集的同时,构建了数据集CH317,将多种特征融合后使用混合增量的方式实现预测[10]。Ding等在伪氨基酸特征下,将模糊K近邻 (Fuzzy K-nearest neighbor,FKNN) 分类器与遗传算法相结合,预测准确率有一定提高[11]。Lin等采用伪氨基酸结合支持向量机方法对蛋白质亚细胞定位进行预测[12]。Zhang等运用支持向量机融合距离频率实现蛋白序列的定位预测[13]。Liao等将伪氨基酸、二肽等多种特征进行融合后通过支持向量机在CH317上取得较好的预测效果[14]。Hu等提取序列之间的网状信息,对位于19个区间的酵母菌数据集进行预测,实现效果较好[15]。Yao等基于序列之间的进化信息,通过位置特异性得分矩阵 (PSSM),统计各氨基酸的突变率取得较好的预测效 果[16]。Liu等提取序列PSSM特征输入SVM进行预测,得到较好的预测效果[17]。Wang等提取序列GO注释信息特征,在支持向量机中实现了革兰氏阴性菌的多区间分类问题[18]。Chen等利用序列的物化属性、PSSM和GO注释3种特征,对多个细菌数据集进行预测,得到较高的预测成功率[19]。总而言之,序列特征越来越丰富,提取过程更复杂,以上所述特征各有优缺点,结合适当的预测分类器可以取得一定的成功率,其中支持向量机、贝叶斯分类器、神经网络等训练较为复杂与耗时。故如何在一般低维或简单特征和低复杂度的预测模型前提下提高识别率是本文重点解决的问题。
KNN是目前理论成熟、应用最为广泛的分类预测算法之一[20],算法简单易优化,这一点对于近年来蛋白序列数量的激剧增长是有利的,但现有研究直接利用KNN进行定位预测效果并不理想,原因在于KNN受模式特征和决策机制影响较大。再考虑到Blast可用于推断结构和功能相似。本文尝试将两者结合起来,改进KNN算法,利用KNN过滤出与预测序列特征相似度较高的序列,再对这些序列进行更精细的Blast比对,作为最终预测依据。具体策略分别以序列的氨基酸组成、二肽和伪氨基酸作为KNN搜索阶段的特征,KNN决策阶段用Blast比对确定蛋白所属定位,最后用Adaboost集成多个KNN子分类器进行定位预测,算法在多个数据集上取得较好的实验效果。文中预测算法通过网站http://www.wsns.org/subloc/homepage_final.jsp实现。
1 材料与方法
1.1 数据集
为了客观评价预测算法的有效性,本文将Chen[10]等使用的CH317作为实验基准数据集。CH317数据集中包含317条蛋白序列,分布在6个位置,其中细胞质蛋白 (Cytoplasmic proteins,cy) 112条,膜蛋白 (Membrane proteins,me) 55条,细胞核蛋白 (Nuclear proteins,nu) 52条,线粒体蛋白 (Mitochondrial proteins,mi) 34条,内质网蛋白 (Endoplasmic reticulum proteins,en) 47条,分泌蛋白 (Secreted proteins,se) 17条。CH317中涉及到的所有蛋白序列均可在uniprot网站下载 (http://www.uniprot.org/)。
除了CH317,为了对预测算法进行进一步评估,本文参考Fan的数据集构建方法[21],具体参数本文不再复述。构建了革兰氏阴性菌数据集 (Gram1253),Gram1253共包含符合规则蛋白序列1 253条,分布于5个位置,如表1 所示。
1.2 序列特征提取
对蛋白序列进行不同特征的提取从而实现蛋白质的亚细胞区间预测是目前研究所采用的主要方法。本文使用氨基酸组成、二肽和伪氨基酸特征。

表1 革兰氏阴性菌数据集分布
1.2.1 氨基酸组成(Amino acid composition,AAC)
不同亚细胞定位中的蛋白质在组成上有很大差别,基于这一特性提出了AAC特征提取方法[22],Nakashima等首次在AAC特征基础上实现了对亚细胞定位的预测[23]。AAC的基本思想:对于任意的蛋白序列,统计构成序列的20种氨基酸各自出现的频率,那么序列的AAC特征可用公式1表示:

上式中,f表示第种氨基酸在序列中出现的频率。
1.2.2 二肽(Dipeptide,Dipe)
二肽特征是基于AAC特征的改进,所谓二肽是指任意2个氨基酸构成的氨基酸对,组成蛋白序列的氨基酸共有20种,因此二肽共有400种,通过统计二肽的频率来描述一条蛋白序列的特征是二肽特征的基本思想[24]。对于任意的序列,其二肽特征可用公式2表示:

上式中,表示第种二肽在序列中出现的频率。
1.2.3 伪氨基酸 (Pseudo amino acid composition,PseAAC)
伪氨基酸特征同样是基于AAC特征的改进,在统计氨基酸频率的基础上,利用维来表示氨基酸之间的位置信息[25]。同AAC特征相比,伪氨基酸特征对序列的刻画更加全面。对于任意的序列,其伪氨基酸特征可用公式3表示:

1.3 预测算法
1.3.1 基于相似性改进KNN分类算法
分类器的设计是对传统KNN算法做改进,修改KNN决策阶段,利用Blast比对取代投票机制确定所属区间位置。
序列相似性常被用来推断结构和功能相似[26],因此,序列比对技术出现在一些区间预测算法中,如将Blast比对作为集成分类器的一个子分类器[27],从Needleman-Wunsch算法的得分矩阵提取特征用于预测[28]。本文采用Blast序列局部比对搜索算法计算蛋白序列之间氨基酸残基的相似比率,从而确定蛋白序列所属位置。通过Blast序列局部比对搜索算法计算得分后,得分最高的蛋白序列便是与检索序列相似度最高的序列。基于改进KNN分类器算法流程见图1。

图1 基于Blast改进的KNN分类算法
文中使用的Blast程序版本为2.2.30,在National Center for Biotechnology Information (NCBI) 官方网站下载 (ftp://ftp.ncbi.nlm.nih. gov/blast/executables/blast+/)。这里采用blastp子程序对蛋白序列的亲缘性进行比对,具体用到的命令及主要参数如下:
1) 数据库格式化
makeblastdb.exe –in DB.fasta-parse_seqids- hash_index–dbtypeprot
其中makeblastdb.exe为格式化数据库命令,–in指定数据库文件,-parse_seqids-hash_index为子序列比对的参数,-dbtype指定比对类型,prot为蛋白序列。
2) 序列比对命令
blastp.exe-task blastp-query que-db DB–out out
使用blastp.exe命令实现蛋白序列比对,-query指定要比对的序列文件,-db为格式化后的数据库文件,–out指定结果输出文件。
1.3.2 Adaboost集成分类预测算法
Adaboost集成分类算法对多个基于Blast改进的KNN分类器进行集成,得到一个较强的分类器[29]。在分类器训练过程中,由于每个分类器的权重都基于前一个分类器的分类效果,因此最后得到的集成分类器效果较好。
给定一个训练数据集={(1,1), (2,2)… (x,y)},其中实例,而实例空间,y属于标记集合{–1,+1},Adaboost的算法流程如下:
步骤1:初始化训练数据的权值分布。每一个训练样本最开始时都被赋予相同的权重:1/N。

步骤2:进行多轮迭代,用=1,2, ...,表示迭代的第轮。
使用具有权值分布D的训练数据集学习,得到基本分类器:

计算G()在训练数据集上的分类误差率
(6)
G()在训练数据集上的误差率e即被G()误分类样本的权值之和。
计算G()的系数,表示G()在最终分类器中的比重

更新训练数据集的权值分布,用于下一轮迭代。
(8)

更新权重使得被基本分类器G()误分类样本的权值增大,而被正确分类样本的权值减小。通过这样的方式,Adaboost方法能“聚焦于”那些较难分的样本上。
其中,Z是规范化因子,使得D+1成为一个概率分布:

步骤3:组合各个弱分类器
(11)
从而得到最终分类器,如下:

Adaboost分类通过对多个相似性比对改进KNN分类器进行集成,实现亚细胞定位预测。一个Adaboost分类器只能完成二分类,所以需要训练多个分类器达到多区间预测,分类器构建过程如下:
1) 对于位于个区间位置的数据集,随机取其中一个区间作为类别1,其余位置作为类别2,转化成一个二分类问题;2)初始化数据集中蛋白序列的权重;3)随机生成值,得到对应的相似性比对改进KNN分类器;4)使用该分类器对数据集进行预测,由预测误差确定分类器系数;5)重复步骤3−4次,得到一个Adaboost分类器;6)根据预测效果更新数据集中样本的权重,用于下一个分类器的训练;7)对于类别2,重复步骤1–6,获取对应的分类器,直到区间无法再细分为止;8)对于个区间的样本,进行–1次训练,得到–1个Adaboost分类器;9)对–1个Adaboost分类器进行集成,得到最终的集成分类器。
1.3.3 评价指标
Jackknife检验是蛋白质亚细胞定位预测中较为常用的检验方法,基本原理为:从数据集中取出一条蛋白序列作为测试序列,剩余序列作为训练集,测试完毕后将该序列放入数据集并取出下一条序列作为测试序列,以此类推直至所有序列预测完毕。本文中的预测算法在Jackknife检验下完成。
参考Chen使用的评价指标,引入敏感性 (S)、特异性 (S)、相关系数 (MMC) 以及总体准确率 ()[10]。
2 结果与分析
KNN分类器值的选取对于整个算法的准确度有很大影响。值越大,包含的蛋白序列数量越多,算法的时间复杂度越高。值越小,则越有可能丢弃掉一些真正有意义的蛋白序列,影响算法的准确度。故文中算法在各数据集的序列预测过程值均取20。
2.1 本文算法在多数据集及多特征下预测结果与分析
基于相似性比对改进KNN的Adaboost集成分类预测算法,对数据集CH317和Gram1253提取3种特征进行预测,CH317的实验结果如表2所示。
由表2可知,除了位置cy和en外,数据集CH317的AAC、PseAAC特征准确率都高于Dipe特征并且总的预测准确率也较高,说明在KNN搜索阶段Dipe特征会误排除一些同模式序列。
基于相似性比对改进KNN的Adaboost集成分类预测算法在数据集Gram1253的实验结果如表3所示。
由表3结果可知,基于相似性比对改进KNN的Adaboost集成分类预测算法在数据集Gram1253上,AAC、PseAAC特征各位置的准确率都高于Dipe特征并且总的预测准确率也较高。总之,与表2的结果一致的是,AAC、PseAAC特征预测准确率都较高,而维数更高的Dipe效果均要差些,带有相邻位置信息的氨基酸对频率不能较准确地代表序列特征。

表2 通过Jackknife检验在数据集CH317上的预测结果

表3 通过Jackknife检验在数据集Gram1253上的预测结果
2.2 本文算法与其他算法预测结果比较
在数据集CH317上,将基于相似性比对改进KNN的Adaboost集成分类预测算法的预测结果同其他方法进行比较,并将结果列于表4中,由于一些其他预测方法不涉及特异性和相关系数,所以这里只对敏感性(S)进行比较。
ID算法使用混合增量 (Increment of diversity, ID) 作为特征进行预测;FKNN使用PseAAC作为特征,结合模糊近邻算法预测;PseAAC_SVM使用PseAAC特征结合支持向量机预测;DF_SVM使用距离频率 (Distance frequency, DF) 结合支持向量机预测;Mix_SVM提出新的PseAAC计算方法结合支持向量机预测;PSSM_SVM使用位置特异性得分矩阵 (PSSM) 结合支持向量机预测。
由表4可以看出,与其他预测算法相比,基于相似性比对改进KNN的Adaboost集成分类预测算法的最高准确率高于其他算法,尤其超过其他以PseAAC为特征的预测算法;在各位置上的预测准确率也较高,特别是CH317的me、se、en位置获得最高准确率,且总体准确率也有一定提高。
为了便于对Gram1253的预测结果进行比较,采用支持向量机作为分类器,分别在3种特征下统计准确率,并与KNN分类器预测结果进行比较,结果列于表5中。

表4 通过Jackknife检验在数据集CH317上不同方法的预测结果
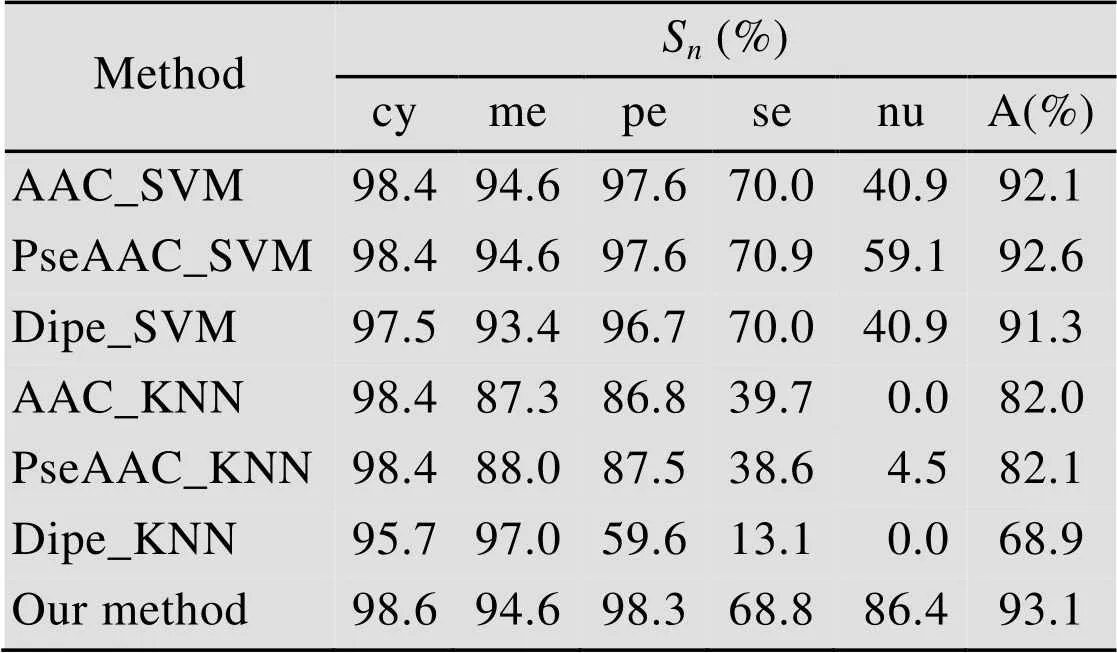
表5 通过Jackknife检验在数据集Gram1253上不同方法的预测结果
由表5可以看出,与支持向量机以及KNN算法相比,当使用AAC特征时Adaboost集成分类预测算法在4个区间cy、me、pe、nu位置的预测效果较好,总体预测准确率有了一定提高。表4、5中,与文中算法预测率接近的是基于支持向量机的预测技术,与它相比,本文算法更适合大数据的处理,算法简单易实现,而支持向量机处理大数据效率低。
3 讨论
蛋白质亚细胞定位预测是生物信息学领域较复杂的研究内容,研究者在序列特征提取与预测算法设计上做了大量工作。在此基础上,不失一般性,本文以常见的AAC、Dipe、PseAAC作为蛋白序列特征,基于相似度高的蛋白序列出现在同一个亚细胞位置中可能性较高的思想构建改进KNN分类器,进而集成改进KNN分类器,实现蛋白质亚细胞定位预测。算法架构可满足大数据处理的要求,对于大数据集,改进KNN分类器便于实现Hadoop等分布式处理架构,缩短算法运行时间。
基于通用性考虑,选用国际公认有效的数据集CH317,并按通用标准构建一个较大数据集用于测试。通过严格的Jackknife检验,数据集CH317和Gram1253在3种特征下最高预测成功率分别为92.4%和93.1%。与一些报道的预测算法相比,集成改进KNN预测算法在3种特征下都取得较好的实验效果,且总体成功率有一定提高,优于直接使用Blast比对预测,说明同源性比对不适合直接用于蛋白质亚细胞定位预测。其中,AAC、PseAAC特征的准确率最为稳定,AAC总体更优,表明在KNN的搜索阶段无需考虑复杂的理化特性。总之,通过在3种特征及多个数据集下的验证测试,集成改进KNN预测算法均取得较好的效果,该算法是一种较为有效的蛋白质亚细胞定位预测算法。
REFERENCES
[1] Cai YD, Liu XJ, Xu XB, et al. Support vector machines for prediction of protein subcellular location. Mol Cell Biol Res Commun, 2000, 4(4): 230–233.
[2] Chou KC, Cai YD. A new hybrid approach to predict subcellular localization of proteins by incorporating gene ontology. Biochem Biophys Res Commun, 2003, 311(3): 743–747.
[3] Chou KC, Elrod DW. Prediction of membrane protein types and subcellular locations. Prot: Struct, Funct, Bioinform, 1999, 34(1): 137–153.
[4] Chou KC, Elrod DW. Protein subcellular location prediction. Protein Eng, Des Select, 1999, 12(2): 107–118.
[5] Reed JC, Paternostro G. Postmitochondrial regulation of apoptosis during heart failure. Proc Natl Acad Sci USA, 1999, 96(14): 7614–7616.
[6] Suzuki M, Youle RJ, Tjandra N. Structure of bax: coregulation of dimer formation and intracellular localization. Cell, 2000, 103(4): 645–654.
[7] Zhou GP, Doctor K. Subcellular location prediction of apoptosis proteins. Proteins: Struct, Funct, Bioinform, 2003, 50(1): 44–48.
[8] Huang J, Shi F. Support vector machines for predicting apoptosis proteins types. Acta Biotheor, 2005, 53(1): 39–47.
[9] Bulashevska A, Eils R. Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinformatics, 2006, 7(1): 298.
[10] Chen YL, Li QZ. Prediction of the subcellular location of apoptosis proteins. J Theoret Biol, 2007, 245(4): 775–783.
[11] Ding YS, Zhang TL. Using Chou’s pseudo amino acid composition to predict subcellular localization of apoptosis proteins: an approach with immune genetic algorithm-based ensemble classifier. Pattern Recognit Lett, 2008, 29(13): 1887–1892.
[12] Lin H, Wang H, Ding H, et al. Prediction of subcellular localization of apoptosis protein using Chou’s pseudo amino acid composition. Acta Biotheor, 2009, 57(3): 321–330.
[13] Zhang L, Liao B, Li DC,et al. A novel representation for apoptosis protein subcellular localization prediction using support vector machine. J Theoret Biol, 2009, 259(2): 361–365.
[14] Liao B, Jiang JB, Zeng QG, et al. Predicting apoptosis protein subcellular location with PseAAC by incorporating tripeptide composition. Protein Pept Lett, 2011, 18(11): 1086–1092.
[15] Hu LL, Feng KY, Cai YD, et al. Using protein-protein interaction network information to predict the subcellular locations of proteins in budding yeast. Protein Pept Lett, 2012, 19(6): 644–651.
[16] Yao YH, Shi ZX, Dai Q. Apoptosis protein subcellular location prediction based on position- specific scoring matrix. J Computat Theoret Nanosci, 2014, 11(10): 2073–2078.
[17] Liu TG, Tao PY, Li XW, et al. Prediction of subcellular location of apoptosis proteins combining tri-gram encoding based on PSSM and recursive feature elimination. J Theoret Biol, 2015, 366: 8–12.
[18] Wang X, Zhang J, Li GZ. Multi-location gram- positive and gram-negative bacterial protein subcellular localization using gene ontology and multi-label classifier ensemble. BMC Bioinformatics, 2015, 16(S12): S1.
[19] Chen J, Xu H, He PA, et al. A multiple information fusion method for predicting subcellular locations of two different types of bacterial protein simultaneously. Biosystems, 2016, 139: 37–45.
[20] Jiang Y, Zhou ZH. Editing training data for kNN classifiers with neural network ensemble[M]//Yin FL, Wang J, GuoCG, Eds. Advances in Neural Networks–ISNN 2004. Berlin Heidelberg: Springer, 2004: 356–361.
[21] Fan GL, Li QZ. Predict mycobacterial proteins subcellular locations by incorporating pseudo- average chemical shift into the general form of Chou’s pseudo amino acid composition. J Theoret Biol, 2012, 304: 88–95.
[22] Nakashima H, Nishikawa K, Tatsuo O. The folding type of a protein is relevant to the amino acid composition. J Biochem, 1986, 99(1): 153–162.
[23] Nakashima H, Nishikawa K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J Mol Biol, 1994, 238(1): 54–61.
[24] Wu C, Whitson G, Mclarty J, et al. Protein classification artificial neural system. Protein Sci, 1992, 1(5): 667–677.
[25] Chou KC, Shen HB. Cell-PLoc: a package of web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc, 2008, 3(2): 153–162.
[26] Nair R, Rost B. Sequence conserved for subcellular localization. Protein Sci, 2002, 11(12): 2836–2847.
[27] Cherian BS, Nair AS. Protein location prediction using atomic composition and global features of the amino acid sequence. Biochem Biophys Res Commun, 2010, 391(4): 1670–1674.
[28] Kim JK, Bang SY, Choi S. Sequence-driven features for prediction of subcellular localization of proteins. Pattern Recognit, 2006, 39(12): 2301–2311.
[29] Lin J, Wang Y. Using a novel Adaboost algorithm and Chou’s pseudo amino acid composition for predicting protein subcellular localization. Protein Pept Lett, 2011, 18(12): 1219–1225.
(本文责编 陈宏宇)
Prediction of protein subcellular locations by ensemble of improved K-nearest neighbor
Wei Xue,Xiongfei Wang,Nan Zhao,Rongli Yang, and Xiaoyu Hong
School of Information Science and Technology, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China
Adaboost algorithm with improved K-nearest neighbor classifiers is proposed to predict protein subcellular locations. Improved K-nearest neighbor classifier uses three sequence feature vectors including amino acid composition, dipeptide and pseudo amino acid composition of protein sequence. K-nearest neighbor uses Blast in classification stage. The overall success rates by the jackknife test on two data sets of CH317 and Gram1253 are 92.4% and 93.1%. Adaboost algorithm with the novel K-nearest neighbor improved by Blast is an effective method for predicting subcellular locations of proteins.
subcellular locations, protein sequence characteristics, K-nearest neighbor, basic local alignment search tool (Blast), Adaboost
Supported by: Fundamental Research Funds for the Central Universities (No. KYZ201668), Natural Science Foundation of Jiangsu Province (No. BK2012363), National Science and Technology Support Program Project (No. 2015BAK36B05).
中央高校基本科研业务费专项资金(No. KYZ201668),江苏省自然科学基金(No. BK2012363),国家科技支撑计划(No. 2015BAK36B05) 资助。
October 18, 2016; Accepted: December 22, 2016
Wei Xue. Tel: +86-21-84396350; E-mail: xwsky@njau.edu.cn
网络出版时间:2017-01-09
http://www.cnki.net/kcms/detail/11.1998.Q.20170109.1240.002.html
猜你喜欢
海洋通报(2022年6期)2023-01-07
海南大学学报(自然科学版)(2020年2期)2020-07-21
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2018年1期)2018-02-02
分析化学(2017年12期)2017-12-25
电子技术与软件工程(2017年14期)2017-09-08
计算机应用(2017年4期)2017-06-27
中成药(2017年3期)2017-05-17
发酵科技通讯(2016年4期)2016-12-21
航天返回与遥感(2014年5期)2014-07-31
