
遗传性凝血因子Ⅴ及Ⅷ联合缺陷症的基因诊断
2016-11-16 09:02汪安友吴竞生孙自敏
安徽医科大学学报 2016年9期
汪安友,刘 欣,吴竞生,孙自敏
遗传性凝血因子Ⅴ及Ⅷ联合缺陷症的基因诊断
汪安友,刘 欣,吴竞生,孙自敏
目的 对2例遗传性凝血因子Ⅴ及Ⅷ联合缺陷症(F5F8D)患者家系进行基因诊断分析。方法 常规凝血功能检测活化部分凝血活酶时间(APTT)、凝血酶原时间(PT)、凝血因子Ⅴ活性(FV:C)及凝血因子Ⅷ活性(FⅧ:C);聚合酶链式反应(PCR)法测序分析LMAN1及MCFD2基因全外显子序列。结果 患者1的APTT及PT分别为78.7 s、21.1 s,FⅧ:C降至23.8%,FV:C为6%;其MCFD2基因3号外显子存在纯合突变,第242碱基位点腺嘌呤突变为胞嘧啶(A>C),导致氨基酸序列第81位天冬氨酸突变成丙氨酸(Asp81Ala)。患者2的APTT及PT分别为78.5 s、19.6 s,FⅧ:C及FV:C分别为24.8%、9.6%;其LMAN1基因12号外显子第1456碱基位点存在鸟嘌呤-胸腺嘧啶-鸟嘌呤纯合缺失(1456delGTG),导致氨基酸序列第486位缬氨酸(Val)缺失。结论 国内首次报道两种新型的MCFD2及LMAN1基因突变,导致两种不同临床表型的遗传性F5F8D。
凝血因子V;凝血因子VIII;LMAN1;MCFD2;突变
遗传性凝血因子V及Ⅷ联合缺乏症(inherited coagulation factor V andⅧdisease,F5F8D)是一种罕见的常染色体隐性遗传性疾病,发病率约为百万分之一,男女均可发病。目前国际上报道的病例主要集中于中东地区。患者临床主要表现为有轻至中度的出血倾向,实验室检测通常为凝血酶原时间(prothrombin time,PT)及凝血活酶时间(activated partial thromboplastin time,APTT)延长,同时伴有凝血因子V及Ⅷ的活性下降,检测后发现多在5%~30%[1-2]。研究[3-4]证实遗传性F5F8D与凝血因子Ⅴ及Ⅷ自身基因无关,主要由细胞内自内质网至高尔基体相关的转运蛋白LMAN1及MCFD2基因突变引起。目前国际上已报道的LMAN1及MCFD2基因不同类型突变导致遗传性F5F8D的共计49种[1-2],国内总共报道过4个完整F5F8D家系研究[5-6],均未发现新型LMAN1及MCFD2基因突变。该研究首次发现2例来自不同家系的疑似遗传性F5F8D患者,对这两个家系做了基因诊断分析,最终确诊了该2例遗传性F5F8D,其基因突变类型均为国内首次发现,现报道如下。
1 材料与方法
1.1 研究对象 家系1资料:先证者,男,30岁,因外伤导致颅内出血就诊于我院急诊,常规凝血功能检测提示APTT、PT均延长,经凝血因子活性检测,临床诊断为凝血因子Ⅴ及Ⅷ联合缺陷,后经基因诊断分析,明确诊断为遗传性F5F8D。先证者父母系近亲结婚,其父亲及大姐为杂合子携带者,其母亲因已病逝,推测为杂合子携带者。家系遗传学图谱见图1。家系2资料:先证者,女,35岁,因不孕症就诊我院门诊,无明显出血倾向,常规凝血功能检测提示APTT、PT均延长,经凝血因子活性检测,临床诊断为凝血因子Ⅴ及Ⅷ联合缺陷,同样经基因诊断分析,明确诊断为遗传性F5F8D。先证者父母系近亲结婚,母亲及姐为杂合子携带者,其父亲因已去逝,推测为杂合子携带者。家系遗传学图谱见图2。
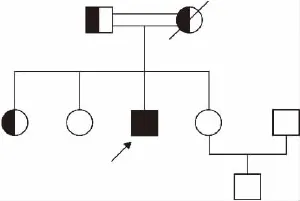
图1 家系1遗传学图谱(箭头代表先证者)
1.2 方法
1.2.1 凝血功能及凝血因子活性检测 0.129 mol/L枸橼酸钠作为抗凝剂,静脉采集血液标本,3 000 r/m离心10 min,分离血浆。采用STAGO公司半自动血凝仪常规检测患者及家系成员外周凝血功能PT、APTT指标。STAGO公司凝血因子检测试剂盒检测分析患者具体凝血因子活性指标。

图2 家系2遗传学图谱(箭头代表先证者)
1.2.2 基因测序分析 0.129 mol/L枸橼酸钠作为抗凝剂,静脉采集患者及其家系成员、健康对照外周血标本,外周血基因 DNA提取试剂盒提取基因DNA。PCR法扩增LMAN1及MCFD2基因所有外显子序列:药物设计参照文献[7],PCR法扩增反应体系:50 μl,Taq buffer 5 μl,25 mmol/L MgCl25 μl,10 mmol/L dNTP 1 μl,10 ng DNA模板1 μl,上下游引物各1 μl,Taq DNA聚合酶5 U/μl 0.5 μl及灭菌蒸馏水35.5 μl。PCR法扩增反应条件:95℃预变性3 min,94℃变性30 s,60℃退火35 s,72℃延伸40 s,10个循环,每个循环退火温度下降0.5℃,94℃变性30 s,55℃退火35 s,72℃延伸40 s,25个循环,72℃延伸3 min。PCR扩增产物经琼脂糖凝胶电泳、割胶纯化后送上海生工测序分析。
2 结果
2.1 实验室凝血功能检测及临床出血表现 通过检测先证者1凝血功能及凝血因子活性发现,其APTT 78.7 s及PT 21.1 s均显著延长,FVIII:C减低为23.8%,FV:C减低为6%,其余凝血因子活性均未见异常;各家系成员的凝血功能指标检测结果及临床出血倾向见表1。
通过检测先证者2凝血功能及凝血因子活性发现,其APTT及PT分别为78.5 s、19.6 s,FⅧ:C降至24.8%,FV:C为9.6%,其余凝血因子活性均未见异常;各家系成员的凝血功能指标检测结果及临床出血倾向见表2。
2.2 基因检测分析 家系1:通过测序检测分析发现先证者1的MCFD2基因所有外显子,分析先证者1在该基因的第3号外显子处检测到1个纯合突变:g.242 A>C,该位点突变直接导致第81位天冬氨酸突变成丙氨酸(Asp81Ala),先证者1的父亲和大姐该位点为杂合突变,50例健康对照均未检测到该突变位点。先证者1及其家系成员的LMAN1基因均未发现异常突变。见图3。
家系2:通过测序检测分析发现先证者2的LMAN1基因所有外显子,分析先证者2在该基因的第12号外显子处检测到1个纯合突变:1456delGTG,该位点纯合缺失突变直接导致第486位缬氨酸丢失;先证者2的母亲和姐姐该位点为杂合缺失突变,50例健康对照均未检测到该突变位点。先证者2及其家系成员的MCFD2基因均未发现异常突变。见图4。

表1 先证者1及其家系成员凝血功能检测及临床出血表现

表2 先证者2及其家系成员凝血功能检测及临床出血表现

图3 家系1成员中MCFD2基因检测发现突变位点

图4 家系2成员中LMAN1基因检测发现突变位点
3 讨论
遗传性F5F8D首次报道于1954年[8],该病在中东地区发病率较高,与这些地区的近亲结婚率高有关。患者通常自幼有轻至中度的出血倾向,包括发生瘀斑、鼻衄、牙龈出血、女性月经过多,以及外伤或拔牙、外科术后过量出血等。实验室检查患者凝血功能指标APTT、PT指标均延长,FV、FⅧ活性水平均降低,多维持在正常人的5%~30%。
目前研究[1]已经证实,该病的分子发病机制与LMAN1或MCFD2基因的突变相关。LMAN1基因位于18q2l,包括13个外显子,编码成熟蛋白由510个氨基酸组成;MCFD2基因位于2p21,较LMAN1基因小,包括4个外显子,其编码成熟蛋白由146个氨基酸组成。LMAN1或MCFD2基因编码的蛋白,可以在细胞内相互结合形成一种稳定的Ca2+依赖转运复合体,主要参与细胞内凝血因子V及Ⅷ从内质网至高尔基体的转运过程。因此,这2个基因的缺陷可以导致细胞内凝血因子V及VIII自内质网至高尔基体的转运障碍,从而使血浆中凝血因子V及Ⅷ水平下降,临床表现为凝血指标的延长,以及不同程度的出血倾向。
本研究中家系1的先证者为30岁男性,因外伤导致颅内出血急诊转入我院,经实验室常规凝血功能检测显示其APTT、PT指标均有不同程度的延长,进一步临床检测了患者相关凝血因子活性,其凝血因子V:C、Ⅷ:C分别降至6%、23.8%。其余凝血因子活性水平均为正常,排除继发性相关因素后,考虑患者父母系近亲结婚,临床诊断考虑为遗传性F5F8D。其父母系表兄妹结婚,该病为常染色体隐性遗传性疾病,对先证者进行LMAN1及MCFD2基因检测,该患者是由MCFD2基因的第3号外显子存在g.242 A>C纯合突变,该位点突变直接导致第81位天冬氨酸突变成丙氨酸(Asp81Ala),先证者的父亲和大姐该位点为杂合突变,先证者母亲因脑出血已病逝,推测其母亲该位点亦为杂合突变。目前国际上已发现18种MCFD2基因突变,其中错义突变仅8种[1,9-11],本课题组最早于2014年在国际上首次报道Asp81Ala位点错义突变,该突变为MCFD2基因上迄今为止发现的第9种类型错义突变[2]。
本研究家系2中的患者为女性,因不孕症就诊,常规检测意外发现凝血功能指标异常,PT、APTT均延长,同样伴有凝血因子V及Ⅷ活性下降。但其自幼无出血倾向,且曾经历妇科手术治疗,追问病史未有术中及术后过量出血表现。经基因检测分析发现:先证者2在LMAN1的第12号外显子处存在1456delGTG纯合突变,直接导致第486位缬氨酸丢失;先证者2的母亲和姐姐该位点为杂合缺失突变,其父亲因去世,无法采集标本检测,结合疾病隐性遗传性规律,推测其父亲该位点同样为杂合缺失突变。目前LMAN1基因的13个外显子已发现36种不同类型突变,这些突变以无义突变或移码突变为主,均可导致LMAN1基因编码蛋白的功能缺失[1,11]。
本研究系在我省首次报道发现2例F5F8D患者,在临床诊疗过程中进行了及时的诊断、分析及治疗,并进一步对患者家系进行基因诊断分析。在国内首次发现2种新型的LMAN1、MCFD2基因突变,这两种类型的基因突变导致2种截然不同的临床表型。尽管先证者1和先证者2检测结果均提示APTT、PT延长;凝血因子Ⅴ、Ⅷ均存在不同程度减少,但是先证者1临床出血倾向较明显,而先证者2并无明显临床出血症状,更有趣的是先证者2甚至在未进行凝血因子替代治疗的情况下,曾经成功经历了妇科手术,术后未见明显出血倾向。不同类型的LMAN1、MCFD2基因突变可以导致不同类型的临床出血表型,这与国外研究[1]报道相一致。至于其机制,很可能是由于LMAN1、MCFD2转运蛋白的不同功能区域,在参与细胞内凝血因子Ⅴ、Ⅷ转运过程中发挥作用大小相关,因此进一步的研究LMAN1、MCFD2转运蛋白的不同结构域的基础功能很有必要,从而为更好的理解认识遗传性F5F8D提供依据。
[1] Zheng C,Zhang B.Combined deficiency of coagulation factorsⅤandⅧ:an update[J].Semin Thromb Hemost,2013,39(6):613 -20.
[2] Wang A,Liu X,Wu J,et al.Combined FV and FⅧ deficiency (F5F8D)in a Chinese family with a novel missense mutation in MCFD2 gene[J].Haemophilia,2014,20(6):e436-8.
[3] Zheng C,Liu H H,Yuan S,et al.Molecular basis of LMAN1 in coordinating LMAN1-MCFD2 cargo receptor formation and ER-to-Golgi transport of FV/FⅧ[J].Blood,2010,116(25):5698-706.
[4] Zhang B,Cunningham M A,Nichols W C,et al.Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex[J]. Nat Genet,2003,34(2):220-5.
[5] 陆晔玲,王学锋,丁秋兰,等.凝血因子Ⅴ及凝血因子Ⅷ联合缺陷患者四例的基因诊断[J].中华检验医学杂志,2010,33(7):618-24.
[6] 葛 菁,薛 峰,顾东生,等.Lman1基因复合杂合突变导致的凝血因子Ⅴ、Ⅷ联合缺乏症[J].中国实验血液学杂志,2010,18 (1):185-90.
[7] Ivaskevicius V,Windyga J,Baran B,et al.The first case of combined coagulation factorⅤand coagulation factorⅧdeficiency in Po land due to a novel p.Tyr135Asn missense mutation in the MCFD2 gene[J].Blood Coagul Fibrin,2008,19(6):531-4.
[8] Oeri J,Matter M,Isenschmid H,et al.Congenital factor V deficiency(parahemophilia)with true hemophilia in two brothers[J]. Bibl Paediatr,1954,58:575-88.
[9] Elmahmoudi H,Wigren E,Laatiri A,et al.Analysis of newly detected mutations in the MCFD2 gene giving rise to combined deficiency of coagulation factors V and VIII[J].Haemophilia,2011,17(5):e923-7.
[10]Hejer E,Adnen L M,Asma J,et al.Identification of a novel mutation in the MCFD2 gene in a Tunisian family with combined factorⅤandⅧdeficiency[J].Tunis Med,2012,90(4):343-4.
[11]Zheng C,Liu H H,Zhou J,et al.EF-hand domains of MCFD2 mediate interactions with both LMAN1 and coagulation factor V or VIII[J].Blood,2010,115(5):1081-7.
Gene diagnosis of hereditary coagulation factor V andⅧ deficiency
Wang Anyou,Liu Xin,Wu Jingsheng,et al
(Dept of Hematology,Affiliated Provincial Hospital of Anhui Medical University,Hefei 230001)
Objective To analyze gene diagnosis of the 2 cases with inherited coagulation factor V andⅧdefects (F5F8D).Methods Routine coagulation function test was used by APTT,PT,FV and FⅧ:C methods;Sequencing analysis all exons of LMANl and MCFD2 genes were detected by PCR method.Results In patient 1,APTT and PT were 78.7s,21.1s;FⅧ:C and FV dropped to 23.8%,6%respectively;the existence of a homozygous mutation of 242 A>C was detected in exon 3 of MCFD2 gene,which led Asp81 mutation to Ala.In patient 2,APTT and PT were 78.5s,19.6s,FV and FⅧ:C were 24.8%,9.6%respectively;the exon 12 of LMAN1 gene had homozygous deletion in 1456GTG,which caused 486Val deletion.Conclusion Two new MCFD2 and LMAN1 mutations are first reported in the world,resulting in two different clinical phenotypes of hereditary coagulation factor V andⅧdeficiency.
coagulation factor V;coagulation factor VIII;LMAN1;MCFD2;mutation
R 554+.9;R 446.9
A
1000-1492(2016)09-1308-04
时间:2016-8-1 14:07
http://www.cnki.net/kcms/detail/34.1065.R.20160801.1407.032.html
2016-05-04接收
安徽省自然科学基金(编号:1308085QH129);安徽高校省级自然科学研究项目(编号:KJ2013Z133);安徽医科大学校科学研究基金项目(编号:2012xkj048)
安徽医科大学附属省立医院血液内科,合肥 230001
汪安友,男,博士,主治医师,责任作者,E-mail:way0623@ 163.com
猜你喜欢
医疗装备(2022年20期)2022-11-11
中国现代医生(2022年19期)2022-11-04
临床输血与检验(2022年3期)2022-06-22
昆明医科大学学报(2022年4期)2022-05-23
浙江医学(2021年4期)2021-01-02
医药前沿(2020年6期)2020-06-30
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
现代检验医学杂志(2016年4期)2016-11-15
重庆医学(2015年12期)2015-03-05
