
酶法合成RNA的相关技术研究进展
2016-10-13 13:20王静潘孝明刘宇梁兴国
生物技术通报 2016年3期
王静 潘孝明 刘宇 梁兴国
(中国海洋大学食品科学与工程学院,青岛 266003)
酶法合成RNA的相关技术研究进展
王静 潘孝明 刘宇 梁兴国
(中国海洋大学食品科学与工程学院,青岛 266003)
近些年来RNA相关研究发展迅速,从质和量两方面对RNA合成技术提出了更高的要求。RNA合成方法包括化学合成和酶法合成两种。目前已商业化的RNA化学合成法能够实现长度小于90个碱基的RNA合成,但合成费用相对较高,使许多研究难以展开。相对于化学合成,酶法合成具有效率高、条件温和的特性,能够合成长序列,是一种高效、低耗的RNA合成方案。酶法合成的技术流程包括转录和加工两步,其中转录的模型主要分为线性转录和滚环转录两种,不同的模型产生的初始转录产物特性不同,需要采用相应的加工方式对其进行处理才能获得准确的目的RNA产物。近年来研究者们不断地研究探索RNA的酶法合成,发现并构建了多种转录模型和初始转录物的加工方案,将从酶法合成RNA的转录模型和加工技术的原理、特点和问题等方面进行概述,以期为后续酶法合成RNA的进一步研究和选择性应用提供参考。
RNA;酶法合成;线性转录;滚环转录;特异位点剪切
随着生物化学与分子生物技术的快速发展,RNA的生物功能被越来越多地发现与报道,如microRNA(miRNA)[1,2]、small interfering RNA(siRNA)[3,4]和nanostructure RNA[5,6]等几类RNA的研究均取得了突破性进展。同时,大量开展RNA相关研究从质和量两方面对RNA的合成技术提出了更高的要求。
RNA材料的合成方式包括化学合成和酶法合成两种。RNA的化学合成是按照3'→5'磷酸二酯键的方向依次连接核苷酸,需采用特定的化学试剂对相应的基团进行适当的加保护和去保护来完成。核酸化学合成起始于20世纪前期[7],并已在30年前实现了自动化,可满足长度小于90个碱基的RNA合成。此外,化学合成可以实现RNA的化学修饰,目前仍是一种广泛应用的RNA合成方法。但由于核糖核苷糖环的2'-OH和3'-OH反应活性近似,导致特定官能团加保护和脱保护困难[8],合成收率随序列长度增加而显著降低[9]。另外,由于化学合成需要采用固相合成法,合成规模难以扩大,无法满足核酸类药物的研发与制备要求,许多报道也提出当前化学合成技术不能满足快速增长的RNA研究的需求[10,11]。
酶法合成RNA是在RNA聚合酶(RNA polymerase,RNAP)体外转录基础上建立的生物制备方法,现已建立了多种模型[12-14]。相对于化学合成的固相合成模式,酶法合成更易于实现较大规模生产,适合进行一些核酸类药物的生产,是一种发展前景良好的RNA合成方法[9,15]。但酶法合成一般较难实现RNA的修饰,仍然有许多技术问题需要解决。深入了解RNA酶法合成相关技术的原理和特性,将有助于上述问题的解决。到目前为止,国内外期刊鲜有酶法合成RNA的相关综述报道。本文就RNA酶法合成的转录模型和加工技术的机制、设计和特点等方面进行概述,以期为后续RNA酶法合成的研究及其应用范围的拓展提供参考。
1 RNA酶法合成简介
酶法合成RNA的制备流程包括转录和加工两个核心部分,转录过程要求合成多拷贝,加工过程要求终产物序列同所需的序列完全一致。转录是酶法合成的基础,具有效率高、反应条件温和的特点,所使用的RNA聚合酶主要包括T7和SP6两种噬菌体RNA聚合酶。在DNA双链模板转录模型中,这两种RNA酶识别各自的启动子,在最适条件下可以获得相对DNA模板几百倍甚至几千倍的RNA转录产物[12,15]。但高效的转录要求起始转录的前三位碱基富含嘌呤,特别是+1位必须是鸟嘌呤[16]。T7和SP6 RNA聚合酶还会在完成模板编码的转录产物合成后继续在其3'端添加1-3 nt任意碱基,造成产物3'端异质[13]。不同的转录模型形成的初始转录物不同,线性转录模型产生的是模板编码的RNA产物单体;滚环模型产生的是模板编码的RNA首尾串联而成的聚合物。因此,要得到目的产物,两种模型转录初始产物都需要在后续加工过程中通过特异性水解酶对其进行切割。本综述分别从转录和加工两个方面分别进行论述。
2 RNA酶法合成中的转录模型
2.1 线性转录模型
根据转录模板的类型,转录大致可以分为线性转录和滚环转录两种,它们具有不同的启动子依赖性、转录效率、特性及问题[12-14]。线性转录模型是指以线性的DNA单链或双链为模板进行转录,根据模型中启动子的存在与否,又可以分为启动子-线性转录模型和无启动子-线性转录模型两类。
最早的启动子-线性转录模型由Melton等[12]在1984年构建,该模型在SP6 RNA聚合酶的作用下能够转录100-6 000个碱基长度的RNA。随着分子生物技术的进步,该方法也得到改进和完善,成为一种常规的转录方案,具体的技术方案如图1所示:(1)将用于转录的双链模板插入到载体的多克隆位点,要求插入片段连接在相应启动子的下游和相应酶切位点之间;(2)重组质粒转化大肠杆菌感受态细胞,测序鉴定筛选阳性克隆,提取阳性克隆的重组质粒;(3)采用限制性内切酶在插入片段的下游切割,使载体线性化;(4)采用T7或SP6 RNA聚合酶进行转录[17-19]。对于双链RNA的制备可以通过将转录模板插入双反向的启动子之间和特异酶切位点之间,进行两向的线性转录而实现。2013年,Rouhana等[20]采用该方法制备了双链RNA,并以此作为材料对涡虫进行RNAi研究。以质粒构建的启动子-线性转录模型已经广泛应用在许多转录或基因表达的相关研究中[21-23],一般涉及的RNA产物较长,对产物末端准确性要求不高,不需要后续的加工处理即可使用。
1987年,Milligan等[13]直接用化学合成的两条分别含有启动子信息和模板信息的DNA链经退火形成双链模板(图2-A),转录合成12-35 nt的RNA,通过优化转录条件最终能够获得毫克级的产物。由于该方法不需要载体的构建和转化等繁琐的操作,所以简单易行,被广泛应用于相对较短的RNA序列的制备[24,25]。
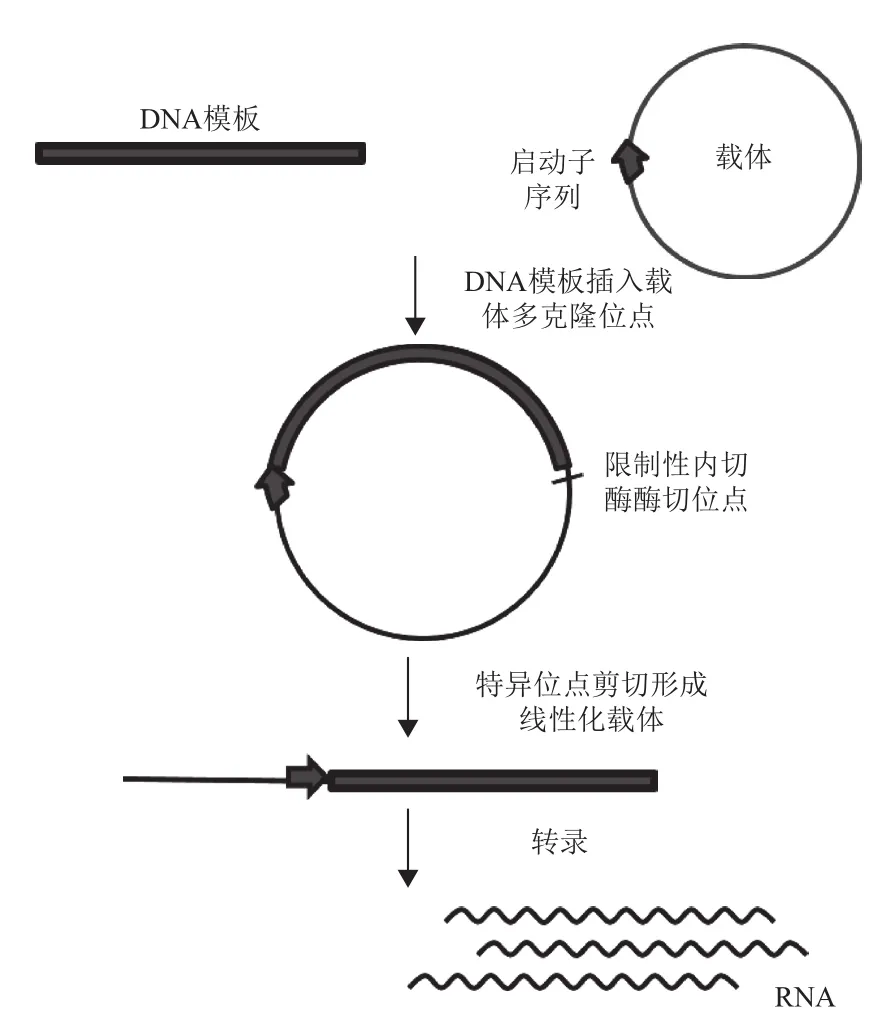
图1 质粒构建启动子-双链转录模型的转录流程
Milligan等[13]还尝试构建了一种截短型启动子-线性转录模型(图2-B),其非模板链只含有T7 RNA聚合酶启动子-17 - +1位的碱基,而模板链与非截短型的(图2-A)相同。结果发现截短型能够形成与非截短型双链模板同样的RNA产物,并且截短的非模板链去除-17 - -14和-4 - +2位一个或数个碱基后仍然可以发生转录。2002年,Donzé和Picard借助该模型建立了一个酶法合成siRNA的方案,并成功地运用到哺乳动物细胞RNAi的研究中[26]。2012年,Cheong等[27]联合该转录模型和DNA-亲和色谱建立了一种快速制备RNA样品的方法,合成的RNA可以满足X-射线、NMR等分析要求。令人意外的是,Sharmeen和Taylor[28]发现了两种无启动子转录模型。其中一种是引物模型(图2-C),由一条短链DNA于模板链的3'端退火,在完全无启动子的情况下引发转录。另一种是无启动子且无引物的单链DNA模板转录模型(图2-D),令人费解的是既没有启动子又没有引物的情况下,SP6 RNA聚合酶能够仅以一条单链DNA寡核苷酸链为模板进行转录,并产生与DNA寡核苷酸链相同大小的RNA。但是这种模型的合成效率显著降低。1988年,Krupp[29]采用不同序列的寡核苷酸链实验该模型的转录特性发现,这种转录可能会引发许多异质产物的产生,不能保证从同一位点起始转录。由此可以看出,在启动子存在的情况下,RNA聚合酶会以启动子转录的机制为主导,而在启动子缺失的情况下,RNA聚合酶可能是识别DNA的二级结构起始转录信号,并以较低的效率完成之后的转录[30,31]。

图2 几种线性转录模型
此外,Milligan等[13]在研究中发现了T7 RNA聚合酶转录产物存在3'端异质的问题,这个缺陷极大影响了酶法合成在RNA合成方面的应用。早期Moran等报道提出DNA模板中引入不能与NTP形成碱基配对的核苷类似物修饰,可能诱导转录在核苷类似物之前终止。他们研究了3种类似物:4-甲基吲哚 β-脱氧核糖核苷(4-methylindole β-deoxynucleoside)、1-萘基 α-脱氧核糖核苷(1-naphthyl α-deoxynucleoside)和1-芘基 α-脱氧核糖核苷(1-pyrenyl α-deoxynucleoside),结果只有4-甲基吲哚β-脱氧核糖核苷在一定程度有所改善,但仍未完全抑制产物异质[24]。1999年,Kao等[32,33]发现DNA模板链的5'端两个碱基进行氧甲基化修饰能够明显降低未端含单个多余碱基的转录产物的产生,但是对含两个多余碱基的产物没有改善。而Sherlin等[34]酶法合成tRNA时对模板链5'端两个碱基进行2'-O-甲基化修饰,获得可用于X-射线晶体分析的RNA样品。可见,对线性转录模型的模板适当修饰可以使初始转录物更接近准确产物,降低后续加工的压力。
2.2 滚环转录模型
1989年,Krupp[12]发现一种SP6和T7 RNA聚合酶的新型转录模型,即滚环转录(Rolling circle transcription,RCT)(图3-A)。该模型以一种单链未成环的DNA(5'-GCATATTTTAAAGTAATATCGTCCA-3')为转录模板,自其3'端第一个C起始转录,到达5'末端后并未终止,而是以DNA的3'端为模板继续转录,形成绕环,这是滚环转录的最初模型。猜测可能的机制是链内部分氢键作用拉近了模板链3'末端与5'末端,形成一个近似的连接端和单链环。1995年,Daubendiek等[35]报道了一类化学合成的单链环状DNA模板的转录模型,图3-B所示为其最初所用的34 nt DNA环状模板,该模板环内无连续互补序列,几乎不可能形成双螺旋的二级结构,最终能够合成12-260次绕环的转录产物。之后,该研究团队探索了不同大小(63 nt/73 nt/83 nt)的环状DNA,发现它们均能够实现转录[31,36]。值得注意的是,这些转录的模板都没有启动子序列。关于单环模板的转录起始机制尚无定论,猜测在缺失启动子的情况下,双链上的突环、泡状结构(bubble)、缺口(gap)和发卡(hairpin)结构都有可能引发转录的起始[30,37,38],DNA单环的构型如何引发RNA聚合酶起始转录还需进一步研究。研究者已将该方法成功地应用到RNA ladder的制作、纳米环载体的构建以及具有催化活性的RNA合成等方面[31,36,39]。2015年,Wang等[40]建立了一种基于滚环模型的RNA合成方法——滚环转录-特异位点断联法(RCT-site-specific disconnection)(图3-C),该方法以目的RNA的互补DNA链为模板,首先以T4 DNA连接酶联合一条短链DNA splint将模板链连接成环,并同时以该splint为引物引发高效率的滚环转录,合成的初始RNA产物是目的RNA的多串联产物,然后经后续特异位点切割处理,形成最终产物。
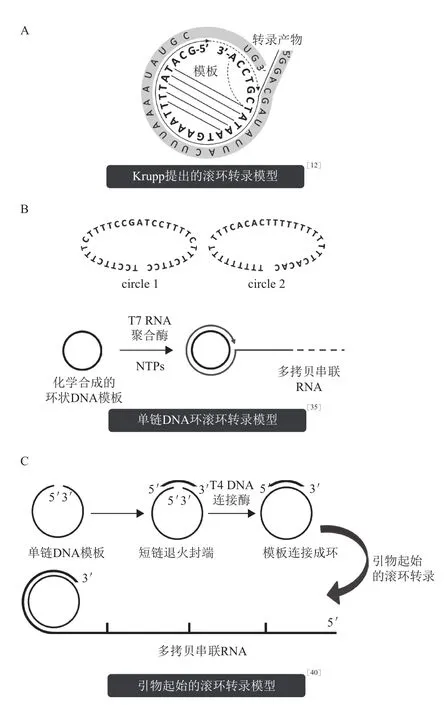
图3 滚环转录模型
3 初始转录物的加工技术
不同的转录模型产生的初始转录物特性不同。对于启动子-线性转录模型,由于其具有转录起始5'端富嘌呤依赖性和3'端产物异质性,往往对模板进行加长设计,转录后形成5'和3'端附含有外序列的初始RNA产物,需将这些多余的序列切除。对于滚环模型,它的初始转录产物是目标RNA的串联产物,需要在RNA单体首尾连接处进行特异位点剪切。所以,大部分初始转录物需用ribozyme、DNAzyme和RNase H等进行酶切处理,以获得准确的RNA产物。
3.1 Ribozyme 特异位点酶切
Ribozyme在酶法合成RNA研究中的应用相对较多。Wichłacz[41]小组为了解决转录终产物3'异质的问题,设计的模板链5'端比目的产物长7 nt,采用trans-acting antigenomic delata ribozyme识别并剪切掉这7 nt多余序列。Price等[42]在转录产物序列前后分别设计了一个hammerhead ribozyme,能够对转录产物两端分别进行自剪切,从而获得准确的RNA产物,并应用到RNA-蛋白复合体的结晶分析。Walker[43]小组在启动子-线性转录模型的基础之上,构建了一个可常规应用的质粒载体,在目的模板的上下游分别插入一个hammerhead ribozyme和一个hepatitis delta virus ribozyme相对应的模板,初始转录产物左右两端的ribozyme分别发挥自剪切功能,形成所需的RNA产物。这种方法需要的模板较长,设计时需考虑较多,不适于大宗的RNA生产。更有意思的是,Daubendiek等[31]在滚环转录模型的应用中将ribozyme设计到环形DNA模板中,获得的串联转录物中的ribozyme能够进行特异位点自剪切而形成ribozyme的单体。
3.2 DNAzyme特异位点酶切
同ribozyme类似,DNAzyme也可以进行特异性剪切。DNAzyme的序列组成包括一个活性中心和两条用于识别底物的结合臂。目前应用于RNA加工处理较多的是8-17和10-23 DNAzyme两种[44-47]。Sohail等[48]用8-17 DNAzyme定点剪切初始转录物5'端多余序列,获得的目的siRNA可成功用于MDAMB-231人类乳腺癌细胞I型胰岛素样生长因子受体(IGF1R)mRNA的RNAi研究。Cheong等[27]借助于DNA亲和色谱建立了一种制备RNA样品的方法,即转录后得到的初始转录物相对于目的产物序列3'端含有一段多余序列,这段多余序列是与亲和色谱柱结合的亲和标签,经过亲和分离纯化后采用10-23 DNAzyme对其剪切加工,再经DNase I水解10-23 DNAzyme获得目的RNA产物。但是,DNAzyme的剪切特性具有序列偏好性且剪切效率差异较大,譬如10-23 DNAzyme偏好剪切5'-嘌呤-嘧啶-3'之间的磷酸二酯键(5'…R↓Y…3',R=A或G,Y=U或C,其中R不形成碱基配对,Y必须与10-23 DNAzyme形成碱基配对),而8-17 DNAzyme主要识别5'…A↓G…3',断裂A与G之间的磷酸二酯键[49]。所以核酶特异位点剪切的加工方法在通用性上有其局限性。
3.3 RNase H 特异位点酶切
目前为止,还没有发现能够识别RNA序列的限制性内切酶。RNase H是一类核酸内切酶,能够水解RNA-DNA杂合子中RNA的磷酸二酯键,而不水解双链RNA或单链RNA。而1987年,Inoue等[50]发现RNase H能识别双链RNA-DNA杂合子中3个连续2'-O-甲基化修饰的核苷酸和连续4个无修饰的核苷酸的序列,从而只切割2'-O-甲基化修饰和无修饰核苷酸之间的磷酸二酯键。之后,氧甲基化修饰的DNA被应用于辅助RNase H特异位点剪切,包括RNA转录产物的加工处理中[51]。Miller等[52]设计转录了一个3'端加长25 nt序列的初始tRNALys,3产物,采用氧甲基化修饰的DNA辅助RNase H对其进行特异酶切,结果证明该方法酶切的产物与化学合成的tRNALys,3大小一致。Wang等[40]对该技术中DNA氧甲基化修饰的参数进行了优化,结合滚环转录和RNase H的特异位点酶切实现了miRNA的准确合成,可得到DNA模板4×103倍以上的miRNA,是目前最具潜力的酶法合成RNA方案之一。
4 结语
高效、准确的RNA合成方式是RNA相关研究的基础和根本,目前已有的转录和加工技术已经能够合成准确的RNA材料,研究者可根据研究需求选择合适的合成方案。相对于化学合成法,酶法合成具有技术门槛低、成本低和反应条件温和等优势,然而,在特殊碱基修饰和序列通用性两个方面仍难以满足研究需求。就目前而言,对于特定碱基的基团修饰基本上只能依赖于化学合成,酶法合成难以实现。酶法合成的通用性相对较低,主要原因在于不同目的RNA的转录起始效率和剪切效率都因序列位点不同而存在较大差异,需要有针对性地进行设计和优化,对于多种RNA的合成负担相对较大。因此,应针对转录和加工过程中的序列反应特性进一步研究。
此外,由于酶法合成需要进行加工处理才能获得准确的RNA产物,相对增加了制备的成本和产物降解的风险,根本问题还是在于T7和SP6 RNA聚合酶的转录特性。2007年,科学家们对一种海洋蓝细菌的噬蓝藻体Syn5进行基因组测序、蛋白结构分析[53,54],成功纯化获得Syn5 RNA聚合酶,2013年报道发现该聚合酶具有较显著的转录优势:(1)对转录初始碱基富嘌呤的偏好性明显低于T7 RNA聚合酶;(2)不具有在准确转录产物3'端添加任意碱基的能力,能够形成准确的转录物[55,56]。这无疑是一项重要的发现,但仍需对其转录的准确性进一步验证。在酶法合成方面,分子技术仍有可能继续改进其高效性和准确性,所以期待更为高效、低耗的酶法合成技术为RNA合成提供新途径,使RNA相关研究得以更广泛地展开。
[1]Llave C, Xie Z, Kasschau KD, et al. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA[J]. Science, 2002, 297(5589):2053-2056.
[2]Cheng AM, Byrom MW, Shelton J, et al. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis[J]. Nucleic Acids Res, 2005, 33(4):1290-1297.
[3]Shen H, Sun T, Ferrari M. Nanovector delivery of siRNA for cancer therapy[J]. Cancer Gene Ther, 2012, 19:367-373.
[4]Lee H, Lytton-Jean A, Chen Y, et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery[J]. Nat Nanotechnol, 2012 7(6):389-393.
[5] Geary C, Rothemund PWK, Andersen ES. A single-stranded architecture for cotranscriptional folding RNA nanostructures[J]. Science, 2014, 345(6198):799-804.
[6] Grabow WW, Jaeger L. RNA self-assembly and RNA nanotechnology[J]. Acc Chem Res, 2014, 47(6):1871-1880.
[7] Michelson A, Todd AR. Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3':5'-internucleotidic linkage[J]. J Chem Soc(Resumed), 1955:2632-2638.
[8] 王来新, 张礼和. RNA化学合成中的保护基[J]. 有机化学,1994, 14(3):242-258.
[9]Gallo S, Furler M, Sigel RK. In vitro transcription and purification of RNAs of different size[J]. Chimia Int J Chem, 2005, 59(11):812-816.
[10] Cai Z, Gorin A, Frederick R, et al. Solution structure of P22 transcriptional antitermination N peptide-box B RNA complex[J]. Nat Struct Biol, 1998, 5(3):203-212.
[11] Hogrefe RI, Midthune B, Lebedev A. Current challenges in nucleic acid synthesis[J]. Isr J Chem, 2013, 53(6-7):326-349.
[12] Melton DA, Krieg PA, Rebagliati MR, et al. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter[J]. Nucleic Acids Res, 1984, 12(18):7035-7056.
[13]Milligan JF, Groebe DR, Witherell GW, et al. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates[J]. Nucleic Acids Res, 1987, 15(21):8783-8798.
[14]Krupp G. Unusual promoter-independent transcription reactions with bacteriophage RNA polymerases[J]. Nucleic Acids Res,1989, 17(8):3023-3036.
[15]Chamberlin M, Ryan T. 4 bacteriophage DNA-dependent RNA polymerases[J]. Enzyme, 1982, 15:87-108.
[16]Chamberlin M, Ring J. Characterization of T7-specific ribonucleic acid polymerase II. Inhibitors of the enzyme and their application to the study of the enzymatic reaction[J]. J Biol Chem, 1973, 248(6):2245-2250.
[17] Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends[J]. Nucl Acids Res, 2003, 31(15):e82.
[18]Brown JA, Bulkley D, Wang JM, et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix[J]. Nat Struct Mol Biol, 2014, 21:633-640.
[19]Brown JA, Valenstein ML, Yario TA. Formation of triple-helical structures by the 3'-end sequences of MALAT1 and MENβnoncoding RNAs[J]. Proc Natl Acad Sci USA, 2012, 109(47):19202-19207.
[20]Rouhana L, Weiss JA, Forsthoefel DJ, et al. RNA interference by feeding in vitro-synthesized double-stranded RNA to planarians:Methodology and Dynamics[J]. Dev Dyn, 2013, 242(6):718-730.
[21]张璐, 钟雄霖, 彭朝晖, 等. 用T7 RNA聚合酶体外转录合成大鼠肝tRNAIle[J]. 生物化学与生物物理进展, 1997, 24(1):78-82.
[22]Pavel I, Belcher A, Browning KS. A method for coupled transcription and aminoacylation of cysteinyl-tRNA[J]. Anal Biochem,2004, 335(2):192-195.
[23]Harrington KM, Nazarenko IA, Dix DB, et al. In vitro analysis of translational rate and accuracy with an unmodified tRNA[J]. Biochemistry, 1993, 32(30):7617-7622.
[24]Moran S, Ren RXF, Sheils CJ, et al. Non-hydrogen bonding‘terminator' nucleosides increase the 3'-end homogeneity of enzymatic RNA and DNA synthesis[J]. Nucleic Acids Res,1996, 24(11):2044-2052.
[25]Höfer K, Langejürgen LV, Jäschke A. Universal aptamer-based real-time monitoring of enzymatic RNA synthesis[J]. J Am Chem Soc, 2013, 135(37):13692-13694.
[26]Donzé O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase[J]. Nucleic Acids Res, 2002, 30(10):e46.
[27]Cheong HK, Hwang E, Cheong C. Rapid preparation of RNA samples using DNA-affinity chromatography and DNAzyme methods[J]. Methods Mol Biol, 2012, 941:113-121.
[28] Sharmeen L, Taylor J. Enzymatic synthesis of RNA oligonucleotides[J]. Nucleic Acids Res, 1987, 15(16):6705-6711.
[29]Krupp G. RNA synthesis:strategies for the use of bacteriophage RNA polymerases[J]. Gene, 1988, 72(1-2):75-89.
[30]Daube SS, von Hippel PH. Functional transcription elongation complexes from synthetic RNA-DNA bubble duplexes[J]. Science, 1992, 258(5086):1320-1324.
[31]Daubendiek SL, Kool ET. Generation of catalytic RNAs by rolling transcription of synthetic DNA nanocircles[J]. Nat Biotechnol,1997, 15(3):273-277.
[32]Kao C, Zheng M, Rüdisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase[J]. RNA, 1999, 5(9):1268-1272.
[33]Kao C, Rüdisser S, Zheng M. A simple and efficient method to transcribe RNAs with reduced 3' heterogeneity[J]. Methods,2001, 23(3):201-205.
[34]Sherlin LD, Bullock TL, Nissan T, et al. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization[J]. RNA,2001, 7(11):1671-1678.
[35]Daubendiek SL, Ryan K, Kool ET. Rolling-circle RNA synthesis:circular oligonucleotides as efficient substrates for T7 RNA polymerase[J]. J Am Chem Soc, 1995, 117(29):7818-7819.
[36] Diegelman AM, Daubendiek SL, Kool ET. Generation of RNA ladders by rolling circle transcription of small circular oligodeoxyribonucleotides[J]. Biotechniques, 1998, 25(5):754-758.
[37] Aiyar SE, Helmann JD, deHaseth PL. A mismatch bubble in double-stranded DNA suffices to direct precise transcription initiation by Escherichia coli RNA polymerase[J]. J Biol Chem,1994, 269(18):13179-13184.
[38]Møllegaard NE, Buchardt O, Egholm M, et al. Peptide nucleic acid. DNA strand displacement loops as artificial transcription promoters[J]. Proc Natl Acad Sci USA, 1994, 91(9):3892-3825.
[39]Diegelman AM, Kool ET. Generation of circular RNAs and transcleaving catalytic RNAs by rolling transcription of circular DNA oligonucleotides encoding hairpin ribozymes[J]. Nucleic Acids Res, 1998, 26(13):3235-3241.
[40]Wang X, Li C, Gao X, et al. Preparation of small RNAs using rolling circle transcription and site-specific RNA disconnection[J]. Mol Ther Nucleic Acids, 2015, 4:e215.
[41]Wichlacz A, Legiewicz M, Ciesiolka J. Generating in vitro transcripts with homogenous 3' ends using trans-acting antigenomic delta ribozyme[J]. Nucleic Acids Res, 2004, 32(3):e39.
[42]Price SR, Ito N, Oubridge C, et al. Crystallization of RNA-protein complexes I. Methods for the large-scale preparation of RNA suitable for crystallographic studies[J]. J Mol Biol, 1995, 249(2):398-408.
[43]Walker SC, Avis JM, Conn GL. General plasmids for producing RNA in vitro transcripts with homogeneous ends[J]. Nucleic Acids Res, 2003, 31(15):e82.
[44]Kasprowicz A, Stokowa-Sołtys K. In vitro selection ofdeoxyribozymes active with Cd2+ions resulting in variants of DNAzyme 8-17[J]. Dalton Trans, 2015, 44(17):8138-8149.
[45]Fokina AA, Stetsenko DA, François JC. DNA enzymes as potential therapeutics:towards clinical application of 10-23 DNAzymes[J]. Expert Opin Biol Ther, 2015, 15(5):689-711.
[46]Robaldo L, Berzal-Herranz A, Montserrat JM, et al. Activity of coremodified 10-23 DNAzymes against HCV[J]. Chem Med Chem,2014, 9(9):2172-2177.
[47]Kumar B, Kumar P, Rajput R, et al. Sequence-specific cleavage of BM2 gene transcript of influenza B virus by 10-23 catalytic motif containing DNA enzymes significantly inhibits viral RNA translation and replication[J]. Nucleic Acid Ther, 2013, 23(5):355-362.
[48]Sohail M, Doran G, Riedemann J, et al. A simple and costeffective method for producing small interfering RNAs with high efficacy[J]. Nucleic Acids Res, 2003, 31(7):e38.
[49]Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme[J]. Proc Natl Acad Sci USA, 1997, 94(9):4262-4266.
[50]Inoue H, Hayase Y, Iwai S, et al. Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H[J]. FEBS Lett, 1987, 215(2):327-330.
[51]Lapham J, Crothers DM. Site-specific cleavage of transcript RNA[J]. Method Enzymol, 2000, 317:132-139.
[52] Miller JT, Khvorova A, Scaringe SA, et al. Synthetic tRNALys,3as the replication primer for the HIV-1HXB2 and HIV-1Mal genomes[J]. Nucleic Acids Res, 2004, 32(15):4687-4695.
[53] Pope WH, Weigele PR, Chang J, et al. Genome sequence, structural proteins, and capsid organization of the cyanophage Syn5:A‘horned' bacteriophage of marine synechococcus[J]. J Mol Biol,2007, 368(4):966-981.
[54]Raytcheva DA, Haase-Pettingell C, Piret JM, et al. Intracellular assembly of cyanophage Syn5 proceeds through a scaffoldcontaining procapsid[J]. J Virol, 2011, 85(5):2406-2415.
[55]Zhu B, Tabor S, Raytcheva DA, et al. The RNA polymerase of marine cyanophage Syn5[J]. J Biol Chem, 2013, 288(5):3545-3552.
[56] Zhu B, Tabor S, Richardson CC. Syn5 RNA polymerase synthesizes precise run-off RNA products[J]. Nucleic Acids Res, 2013, 42(5):e33.
(责任编辑 狄艳红)
Research Progress on Enzymatic Synthesis of RNA
WANG Jing PAN Xiao-ming LIU Yu LIANG Xing-guo
(College of Food Science and Engineering,Ocean University of China,Qingdao 266003)
With the rapid development of RNA related studies,RNA synthesis technology is further challenged from two aspects of quality and quantity. At present there are mainly two methods for RNA synthesis,chemical synthesis and enzymatic synthesis. By the commercialized chemical way,the synthesis of RNA with < 90 bases is achieved;however,the cost of the synthesis is relatively high,which leads the initiation of many researches in difficulty. Compared to the chemical way,the enzymatic synthesis presents the characteristics of the high efficiency and mild reaction conditions,and by it a long sequence of RNA can be synthesized,thus,it is an efficient and lowcost measure for RNA synthesis. The enzymatic production of RNA is in two steps,transcription and processing of initial transcripts. The transcription may be further classified into the linear transcription and the rolling circle transcription model,with which the characteristics of initial transcripts vary,and the corresponding processing methods should adapted to the varied initial transcripts,thereby,the accurate target RNA products may be obtained. Recently,as the exploration of enzymatic synthesis continues,many novel transcription and processing strategies have been developed. In this review,the mechanisms,characteristics and problems of the transcription models and processing methods in enzymatic synthesis were summarized,aiming at providing the
for further study and selective utilization of enzymatic synthesis of RNA.
RNA;enzymatic synthesis;linear transcription;rolling circle transcription;site-specific cleavage
10.13560/j.cnki.biotech.bull.1985.2016.03.008
2015-05-19
国家自然科学基金项目(31201327,31301420)
王静,女,博士,研究方向:核酸化学与生物技术;E-mail:wodejia_sun@126.com
梁兴国,男,博士,研究方向:核酸化学与生物技术;E-mail:liangxg@ouc.edu.cn
猜你喜欢
海外星云 (2021年21期)2021-01-19
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
科学24小时(2019年5期)2019-06-11
发明与创新(2019年9期)2019-03-26
国外医药(抗生素分册)(2016年3期)2016-07-12
当代经济(2016年26期)2016-06-15
中国酿造(2016年12期)2016-03-01
中国粮油学报(2016年5期)2016-01-23
中成药(2014年11期)2014-02-28
