
大鼠ASIC1基因启动子荧光素酶报告质粒的构建及其功能鉴定
2016-09-10 09:14周仁鹏吴小山王志森谢亚亚代贝贝王志强葛金芳陈飞虎
安徽医科大学学报 2016年5期
周仁鹏,吴小山,王志森,谢亚亚,李 悦,代贝贝,王志强,葛金芳,陈飞虎
大鼠ASIC1基因启动子荧光素酶报告质粒的构建及其功能鉴定
周仁鹏,吴小山,王志森,谢亚亚,李悦,代贝贝,王志强,葛金芳,陈飞虎
目的 构建大鼠酸敏感离子通道1(ASIC1)基因启动子荧光素酶报告质粒pGL3-ASIC1-promoter,并进行功能的鉴定。方法 设计、合成ASIC1启动子引物,采用聚合酶链反应(PCR)技术从大鼠全基因组DNA中扩增出ASIC1启动子片段;Nhe I和Xho I双酶切后将目的片段连接到pGL3-Basic报告载体上;构建的pGL3-ASIC1-promoter重组质粒和pRLTK内参质粒瞬时共转染293T细胞检测ASIC1启动子活性。结果 PCR扩增得到大鼠ASIC1基因启动子片段;成功构建pGL3-ASIC1-promoter报告基因载体,菌落PCR和测序结果表明启动子DNA序列正确。与空质粒pGL3-Basic转染组相比,pGL3-ASIC1-promoter质粒转染组的荧光素酶活性明显增加(P<0.01)。结论 成功构建了大鼠ASIC1基因启动子报告基因载体,为探究ASIC1转录表达的调控机制奠定基础。
ASIC1;启动子;荧光素酶;报告基因
网络出版时间:2016-4-19 11:04:48 网络出版地址:http://www.cnki.net/kcms/detail/34.1065.R.20160419.1104.006.html
酸敏感离子通道(acid sensing ion channels,ASICs)是一类胞外H+激活的阳离子通道,属于阿米洛利敏感的上皮钠通道/退变素(epithelial Na+channels/degenerin,ENaC/DEG)超家族,广泛地表达在中枢和外周神经系统以及非神经组织中,通过介导Na+和Ca2+内流参与各种病理生理效应[1]。ASIC1因其对Ca2+有通透性而有广泛的生物学功能及重要的生理病理学意义[2]。ASIC1a在佐剂性关节炎(adjuvant arthritis,AA)大鼠关节软骨细胞上有表达[3-4],并参与关节软骨的代谢过程[5];阻断ASIC1a可以降低酸诱导的AA大鼠关节软骨细胞凋亡[6-7]。由此推测ASIC1可能在类风湿关节炎(rheumatoid arthritis,RA)的发生和发展过程中发挥着重要作用。该研究通过构建大鼠ASIC1启动子报告基因质粒,并观察其转录调控活性,为探究其基因转录活性的调控机制以及为进一步探究ASIC1在RA中的作用奠定基础。
1 材料与方法
1.1材料 293T细胞株(本实验室保存);DMEM/ F-12培养基(美国Hyclone公司);胎牛血清(美国Gibco公司);琼脂糖凝胶DNA回收试剂盒、大肠杆菌感受态细胞 DH5α、Taq酶和 dNTP、PrimeSTAR、6 ×Loading Buffer、DL2000 DNA Marker(日本TaKaRa公司);1 000 bp DNA ladder Marker(美国Thermo Scientific公司);细胞/组织基因组DNA提取试剂盒(美国 Axygen公司);限制性内切酶Nhe I和Xho I(美国NEB公司);pGL3-Basic载体、内参质粒pRLTK、Dual-Luciferase®Reporter Assay Kit、小抽试剂盒、GloMax®Multi Jr单管型多功能检测仪(美国Promega公司);无缝克隆试剂盒(上海生博生物医药科技有限公司);脂质体Lipofectamine2000、Opti-MEM(美国Invitrogen公司)。
1.2方法
1.2.1基因组DNA提取 采用细胞/组织基因组DNA提取试剂盒抽提大鼠脑组织DNA,具体实施步骤按照产品操作说明手册进行。对提取DNA测量光密度(optical density,OD)值后,加ddH2O保存于-20℃。
1.2.2ASIC1基因启动子序列PCR扩增及纯化登陆美国国立生物技术信息中心(national center for biotechnology information,NCBI),搜索GenBank数据库中的大鼠ASIC1 DNA序列(Gene ID:79123),利用Primer 5.0软件辅助设计针对ASIC1基因启动子区(-1545~+384)的引物,上游引物:5′-CTTACGCGTGCTAGCGCTTTATAGGTCGGCCATGGA-3′;下游引物:5′-ATCGCAGATCTCGAGCCTTGCCAAGGGGATCCTG-3′,上、下游引物分别引入酶切位点分别为Xho I、Nhe I,实验所需引物均委托上海捷瑞生物工程有限公司进行合成。以所提取的大鼠脑基因组DNA为扩增模板,采用50μl反应体系:前段引物(10μmol/L)1.0μl、后段引物(10μmol/L)1.0μl、5×Buffer(with Mg2+)10μl、dNTP(各2.5 mmol/L)4μl、DNA模板1.0μl、PrimeSTAR 0.5μl,加ddH2O补足至50μl。循环条件为98℃预变性3 min;98℃变性10 s、55℃退火15 s、72℃延伸1 min,循环30次;72℃10 min终止反应。PCR产物进行10 g/L琼脂糖凝胶电泳,并对PCR扩增产物回收纯化备用。
1.2.3pGL3-ASIC1-promoter载体的构建 用Xho I和Nhe I分别双酶切PCR产物和pGL3-Basic载体,回收目的片段。将线性化载体和回收的目的片段以1:2(摩尔比)的比例加到试管中进行重组反应(42℃孵育30 min),放置2~3 min后取10μl反应液体转化到大肠杆菌DH5α感受态细胞中,并涂布于含氨苄青霉素(100 mg/L)抗性的LB琼脂平板上,37℃培养16 h后,从转化子的平板上随即挑取8个菌落,挑取菌落接种于10μl含氨苄抗性的LB培养液中。取培养后的菌液(1μl),进行菌落PCR,上游引物:5′-TCGCAGACACTCGTAGGCG-3′;下游引物:5′-CCTTATGCAGTTGCTCTCC-3′,以培养后的菌液为模板,采用20μl反应体系:前段引物(10 μmol/L)0.8μl、后段引物(10μmol/L)0.8μl、5× Buffer(with Mg2+)2μl、dNTP(各 2.5 mmol/L)1.6 μl、DNA模板1.0μl、Taq酶0.1μl,加ddH2O补足至20μl。循环条件为94℃预变性5 min;94℃变性30 s、55℃退火30 s、72℃延伸1 min,循环30次;72℃10 min终止反应。进行凝胶电泳、观察并照相。选取阳性克隆接种到LB液体培养基,37℃摇菌过夜;取含ASIC1启动子的菌液行DNA测序,之后提取质粒备用。构建携带ASIC1基因启动子序列的pGL3表达载体,命名为pGL3-ASIC1-promoter。
1.2.4荧光素酶报告基因载体转染239T细胞293T细胞于含10%胎牛血清(fetal bovine serum,FBS)的DMEM/F-12培养基中进行培养,转染前24 h将对数生长期的293T细胞消化计数(密度为1× 105个/孔),种于24孔板中,置于37℃、5%CO2培养至60%~70%,用50μl Opti-MEM培养基稀释0.9μg质粒报告质粒和0.1μg pRL-TK质粒,同时1μl Lipofectamine2000溶于50μl Opti-MEM培养基中,5 min后将两者混合室温静置20 min;用PBS清洗细胞3次后加入400μl Opti-MEM,再逐滴加入质粒-脂质体混合物,轻轻摇晃,置37℃、5%CO2培养6 h,然后更换含10%FBS的DMEM/F-12培养液500μl继续培养48 h。实验中将作为阴性对照的pGL3-Basic空载体与作为内参的pRL-TK质粒共转染。每组试验重复3次,试验设6个复孔/次。
1.2.5荧光素酶报告基因活性的检测 细胞转染48 h后,弃培养液,PBS清洗细胞1次,室温摇动15 min。按照双荧光素酶检测试剂盒的操作说明书,取100μl Luciferase Assay Buffer II于1.5 ml的离心管中;将GloMax®Multi Jr单管型多功能检测仪测定间隔设定为2 s,测读为10 s;将293T细胞裂解液转移到新的1.5 ml离心管中,用移液器轻轻吸打2~3次混匀,切勿要旋涡振荡,将离心管放入到化学发光仪中读取萤火虫荧光素酶发光值;再将离心管移出发光仪,加入100μl Stop&Glo Reagent,用移液器吹打混匀后,再将离心管放回多功能检测仪中读取海肾荧光素酶发光值,计算前者与后者的发光值的比值,即为被检测质粒的相对荧光素酶活性(relative light unit,RLU)。
1.3统计学处理 采用SPSS 16.0统计软件进行分析,数据以¯x±s表示,应用t检验行两组间的差异性分析,以α=0.05作为检验水准。
2 结果
2.1pGL3-Basic载体双酶切 用NheⅠ和XhoⅠ对pGL3-Basic表达载体(图 1A)进行酶切,琼脂糖电泳结果表明,在4 000~5 000 bp有明显条带,回收4 807 bp长度的pGL3-Basic载体片段(图1B)。
2.2大鼠ASIC1基因启动子的扩增 以基因组DNA为模板扩增ASIC1启动子序列,扩增的 PCR产物经过1.2%琼脂糖电泳后,结果可见在约2 000 bp处出现单一的明亮的条带,与1 929 bp的 ASIC1启动子片段大小相符合(图2)。
2.3大鼠ASIC1基因启动子荧光素酶报告质粒的构建与鉴定 PCR扩增大鼠ASIC1基因启动子片段(-1545~+384)后,将其克隆至pGL3-Basic载体上构建 pGL3-ASIC1-promoter质粒。重组质粒经转化后涂布于LB琼脂平板上,挑取8个克隆摇菌,菌落PCR鉴定结果显示均出现了阳性克隆片段(图3A);另外将所得阳性克隆送公司测序,DNA测序结果显示,两者序列一致(图3B)。上述结果提示大鼠pGL3-ASIC1-promoter质粒已成功构建。
2.4大鼠ASIC1基因启动子荧光素酶报告质粒的功能鉴定重组质粒 pGL3-ASIC1-promoter和pGL3-Basic质粒分别与内参质粒pRL-TK共转染293T细胞后,观察萤火虫以及内参海肾荧光素酶报告基因活性,两者活性的比值即为ASIC1启动子活性,内参质粒pRL-TK可以排除由于转染效率和细胞数目等因素不同所引起的组间差异,结果表明,与空质粒pGL3-Basic转染组相比,重组质粒pGL3-ASIC1-promoter质粒转染组的荧光素酶活性高14倍,两者相比,差异有统计学意义(t=-19.077,P<0.01)(图4)。提示构建的大鼠pGL3-ASIC1-promoter质粒可以反映ASIC1启动子的功能。
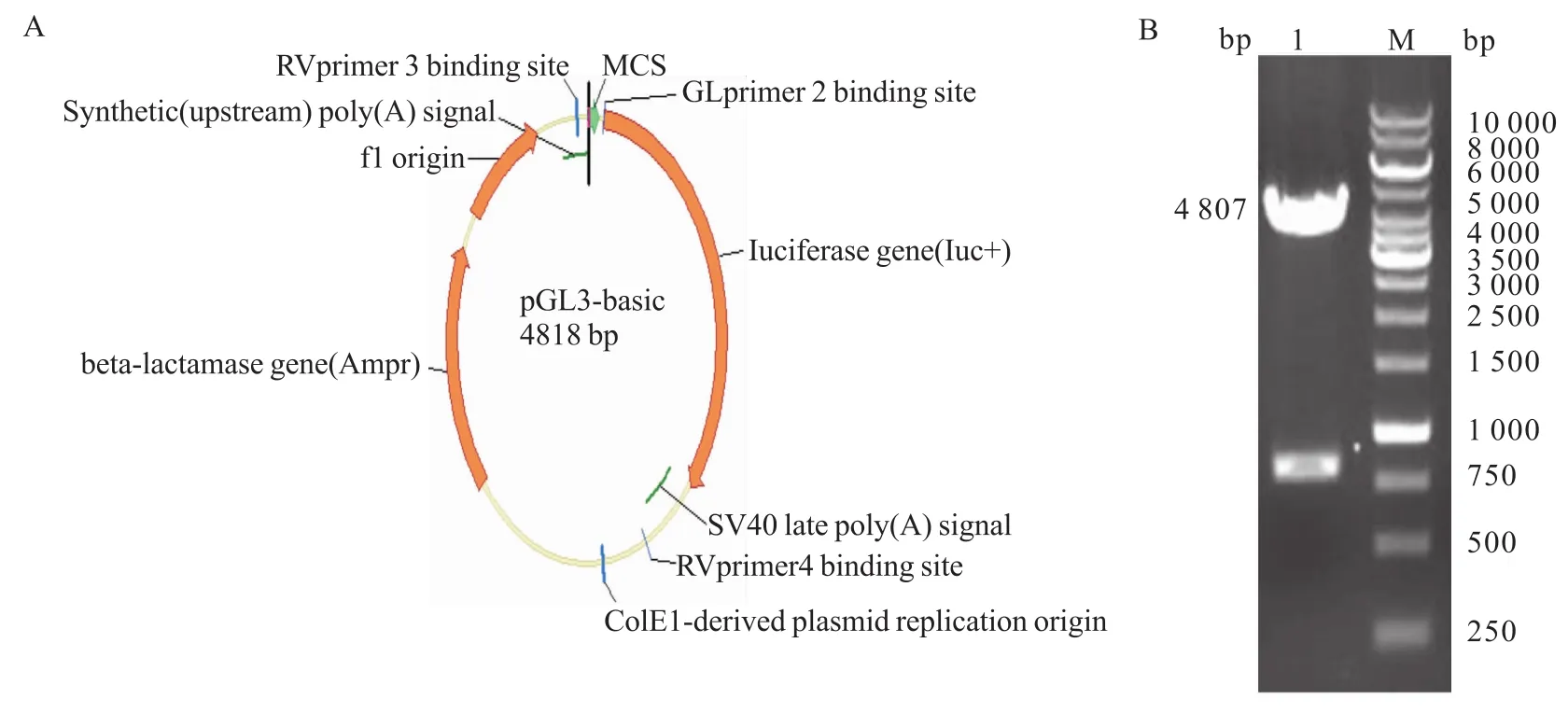
图1 载体及双酶切琼脂糖电泳A:pGL3-Basic质粒载体图谱;B:NheⅠ、XhoⅠ双酶切后结果;M:Marker;1:4 807 bp的 pGL3-Basic片段
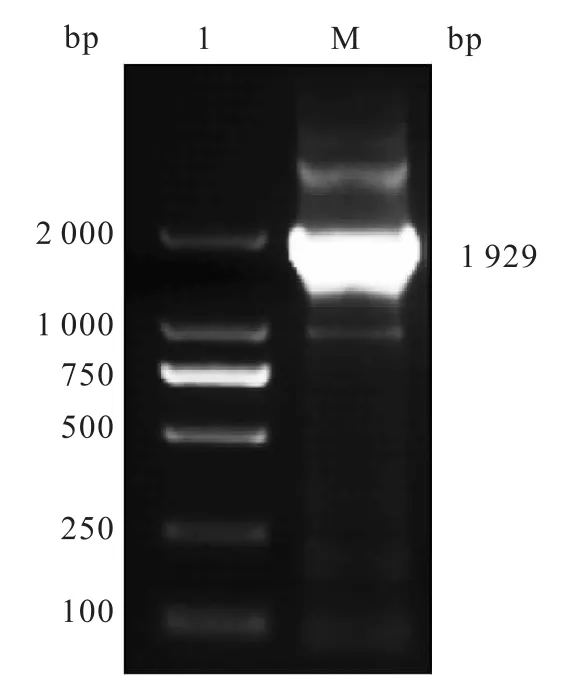
图2 ASIC1启动子PCR扩增结果M:Marker;1:1 929 bp的 ASIC1启动子片段
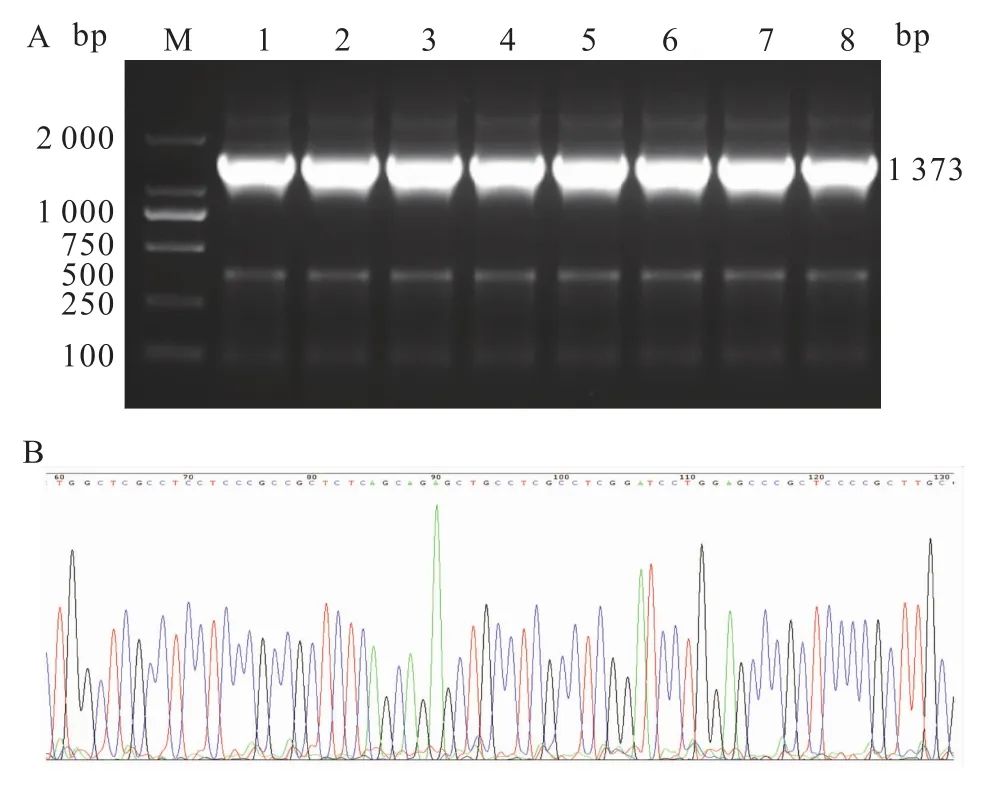
图3 pGL3-ASIC1-promoter质粒的鉴定A:菌落PCR扩增结果;M:Marker;1~8:8个克隆的 pGL3-ASIC1-promoter质粒菌落PCR扩增产物;B:pGL3-ASIC1-promoter质粒的测序图谱(部分)
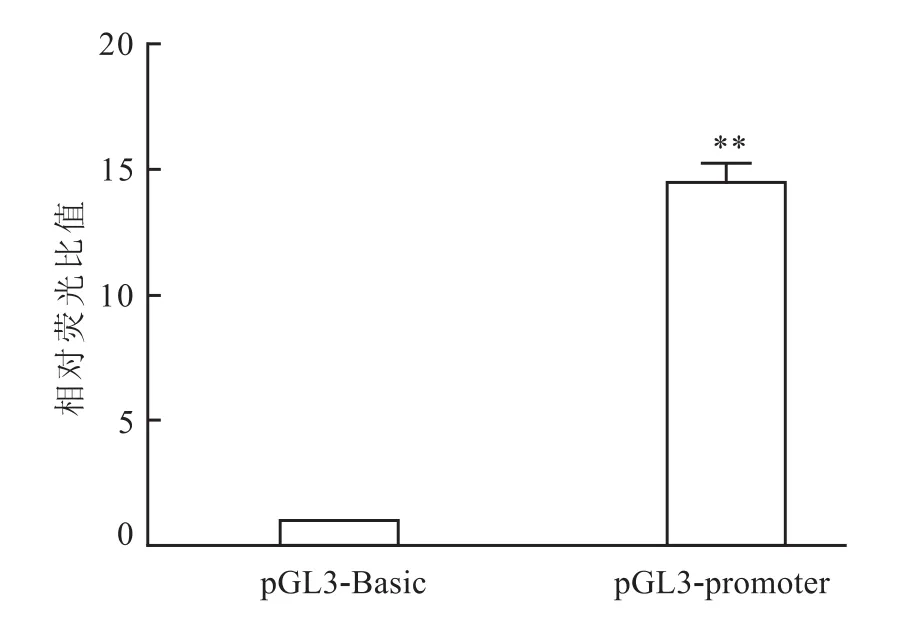
图4 pGL3-ASIC1-promoter转染293T细胞后的相对荧光素酶活性与pGL3-Basic组比较:**P<0.01
3 讨论
ASICs是一类H+门控的阳离子通道,当细胞外pH值下降(H+浓度上升)时,ASICs被激活,其离子通道打开,通过对Na+和Ca2+的通透形成内向电流,引起细胞膜去极化而产生兴奋性效应;几乎所有疾病如癌症、炎症、缺血、缺氧等的过程都会引起不同程度的pH值下降,而ASICs作为一种重要的酸感受器,感受这种变化进而影响着组织病理生理的改变[8]。近年来对ASICs的研究发展非常迅速,到目前为止,已克隆出来由4个基因编码(ASIC1/Ac-cn2、ASIC2/Accn1、ASIC3/Accn3、ASIC4/Accn4)的7个 ASIC亚基:ASIC1a、ASIC1b、ASIC1b2、ASIC2a、ASIC2b、ASIC3、ASIC4;其中ASIC1a因其可以参与Ca2+内流的调控并具有重要的生物学特性和病理生理学价值而深受关注[9]。大鼠ASIC1基因的全长约为28 620 bp,包括由13个外显子和12个内含子,定位于第7号染色体7q36;其mRNA约3 800 bp,基因编码528个氨基酸,蛋白质分子量约59 ku。近些年来,已有对ASIC1基因敲除、转录后水平调控及下游靶分子的研究[10-11],但对于其转录水平调控研究较少。启动子是转录激活过程中起动作用的特异的DNA序列,与序列上的转录因子结合后可以调节基因的转录,进而影响蛋白质翻译,因此对于ASIC1基因启动子研究显得尤为必要。
目前,荧光素酶报告基因是检测检测转录因子与目的基因启动子区DNA相互作用的一种检测方法;即将目的基因的启动子序列插入到报告基因质粒中,然后将构建的质粒转入相应的细胞中,最后通过观察报告基因表达的水平,可反映插入的启动子序列的功能[12-13]。在前期研究的基础上,本实验运用PCR技术扩增了ASIC1启动子序列,并将这段序列定向克隆入pGL3-Basic载体中,菌落PCR和DNA测序分析证明导入序列、片段大小正确,成功构建了ASIC1的报告基因质粒。将构建成功的重组质粒pGL3-ASIC1-promoter与内参质粒pRL-TK共转染到293T细胞中,采用双荧光素酶报告基因(DLRTM)检测系统进行相对表达活性的检测。结果显示,空载体pGL3-Basic阴性对照组的转录活性非常的低;而pGL3-ASIC1-promoter质粒转染组有明显的转录活性。证实了1 929 bp碱基ASIC1基因启动子区启动了荧光素酶的表达,提示这段靶序列具有较好的功能。成功构建出大鼠pGL3-ASIC1-promoter质粒是探究ASIC1表达转录的前提,这为进一步寻找ASIC1基因调控区中的顺式作用元件及其转录因子、甲基化状态提供了有效工具,也为探究和筛选ASIC1a的抑制剂奠定了前期基础。
[1] LiW G,Xu T L.Acid-sensing ion channels:a novel therapeutic target for pain and anxiety[J].Curr Pharm Des,2015,21(7):885-94.
[2] Li X,Wu FR,Xu R S,etal.Acid-sensing ion channel1a-mediated calcium influx regulates apoptosis of endplate chondrocytes in intervertebral discs[J].Expert Opin Ther Targets,2014,18(1):1-14.
[3] Yuan F L,Chen FH,Lu W G,etal.Acid-sensing ion channel 1amediates acid-induced increases in intracellular calcium in rat articular chondrocytes[J].Mol Cell Biochem,2010,340(1-2):153-9.
[4] 袁凤来,陈飞虎,李 霞,等.大鼠关节软骨酸敏感离子通道的表达[J].安徽医科大学学报,2007,42(5):513-6.
[5] Yuan F L,Chen F H,Lu W G,et al.Inhibition of acid-sensing ion channels in articular chondrocytes by amiloride attenuatesarticular cartilage destruction in rats with adjuvant arthritis[J].Inflamm Res,2010,59(11):939-47.
[6] Hu W,Chen F H,Yuan F L,etal.Blockade of acid-sensing ion channels protects articular chondrocytes from acid-induced apoptotic injury[J].Inflamm Res,2012,61(4):327-35.
[7] Rong C,Chen F H,Jiang S,et al.Inhibition of acid-sensing ion channels by amiloride protects ratarticular chondrocytes from acidinduced apoptosis via amitochondrial-mediated pathway[J].Cell Biol Int,2012,36(7):635-41.
[8] 周仁鹏,陈飞虎.酸敏感离子通道在类风湿关节炎中作用的研究进展[J].中国药理学通报,2015,31(3):315-8.
[9] Deval E,Lingueglia E.Acid-Sensing Ion Channels and nociception in the peripheraland central nervous systems[J].Neuropharmacology,2015,94:49-57.
[10]Yu XW,Hu Z L,Ni M,et al.Acid-sensing ion channels promote the inflammation andmigration of cultured ratmicroglia[J]. Glia,2015,63(3):483-96.
[11]周仁鹏,吴小山,王志森,等.ASIC1基因敲除小鼠的繁殖及基因鉴定[J].安徽医科大学学报,2015,50(9):1341-3.
[12]Tirodkar T S,Lu P,Bai A,et al.Expression of ceramide synthase 6 transcriptionally activates acid ceramidase in a c-Jun N-terminal kinase(JNK)-dependentmanner[J].JBiol Chem,2015,290(21):13157-67.
[13]赵 虹,高星杰,葛 林,等.人Tudor-SN基因启动子荧光素酶报告基因表达质粒的构建及活性检测[J].医学分子生物学杂志,2011,8(3):198-203.
Construction and identification of the rat ASIC1 promoter luciferase reporter p lasm id
Zhou Renpeng,Wu Xiaoshan,Wang Zhisen,et al
(School of Pharmacy,Anhui Medical University,Hefei 230032)
Objective To construct ratacid sensing ion channel1(ASIC1)promoter recombined luciferase reportergene plasmid,and then identify its function.Methods Primers were designed and synthetised,ASIC1 promoter fragment from rat genome DNA was replicated by polymerase chain reaction(PCR).The luciferase report gene pGL3-Basic reporter vector and ASIC1 promoter were digested with restriction enzymes NheI and XhoI separately,and then ASIC1 promoter was connected to pGL3-Basic reporter vector.293T cells were transiently co-transfected with the constructed pGL3-ASIC1-promoter plasmid and pRL-TK control plasmid,and then detected for luciferase activity after 48 hours.Results Rat ASIC1 gene promoter was amplified by PCR and pGL3-ASIC1-promoter reporter vector was successfully constructed,and the result of colony PCR and sequencing analysis of recombined plasmid were correct.The transcriptional activity of pGL3-ASIC1-promoter plasmid group was significantly increased compared to that of pGL3-Basic plasmid group(P<0.01).Conclusion The rat ASIC1 promoter luciferase reporter gene vector can be successfully constructed,which provides a pivotal basis for further study of regulatorymechanism of ASIC1 gene in transcription.
ASIC1;promoter;luciferase;reporter gene
R 392.2
A
1000-1492(2016)05-0620-05
2016-03-02接收
国家自然科学基金(编号:81271949)
安徽医科大学药学院,合肥 230032
周仁鹏,男,博士研究生;
陈飞虎,男,博士生导师,责任作者,E-mail:cfhchina@sohu.com
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
江西农业学报(2021年4期)2021-04-20
中日友好医院学报(2021年1期)2021-04-14
山东医药(2020年9期)2020-05-20
中国药理学通报(2015年2期)2016-01-12
热带农业科学(2015年9期)2015-10-14
西南医科大学学报(2015年1期)2015-08-22
中国医学科学院学报(2015年5期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年27期)2015-02-28
