
猪伪狂犬病病毒套式PCR检测方法的建立与应用
2016-07-16 02:30吴旭锦朱小甫
动物医学进展 2016年6期
吴旭锦,朱小甫
(咸阳职业技术学院畜牧兽医研究所,动物疫病分子生物学诊断实验室,陕西咸阳 712000)
猪伪狂犬病病毒套式PCR检测方法的建立与应用
吴旭锦,朱小甫*
(咸阳职业技术学院畜牧兽医研究所,动物疫病分子生物学诊断实验室,陕西咸阳 712000)
摘要:对伪狂犬病病毒(PRV)国标PCR方法进行优化,建立一种高灵敏度的套式PCR方法。根据伪狂犬病病毒gD基因序列,设计并合成了2对引物,通过反应体系和条件的优化建立套式PCR方法。通过灵敏性试验、特异性试验、病料检测对比试验等验证建立方法的适用性。结果表明,建立的方法检测伪狂犬病病毒DNA的极限为2.6×10-7ng/mL,灵敏度比国家标准方法提高了1 000倍,从疑似PRV感染组织病料中能大幅度提高阳性检出率。本研究成功建立了一种灵敏度高、特异性好的快速检测猪伪狂犬病病毒的套式PCR检测方法。
关键词:伪狂犬病病毒;套式PCR;诊断;应用
猪伪狂犬病(Porcine pseudorabies,PR)是由伪狂犬病病毒(Pseudorabies virus,PRV)引起的一种以感染猪发热、脑脊髓炎为特征的传染病,PRV感染怀孕母猪引起流产、死胎,初生仔猪感染病死率高,成年猪多呈隐性感染,是PRV传播的危险传染源[1]。PR在我国猪群中流行范围很广,造成巨大的经济损失[2-3]。20世纪70年代以来,伪狂犬病弱毒疫苗以及基因缺失苗的广泛应用,一度使我国伪狂犬病得到了有效控制。但近几年来,猪伪狂犬病发病报道激增,呈现卷土重来之势,给养猪业造成沉重打击[4-6]。为防控PR,特异灵敏的诊断手段是必不可少的。我国国家标准《伪狂犬病诊断技术(GB/T 18641-2002)》中提出的诊断技术手段包括病毒分离鉴定、聚合酶链反应(PCR)、家兔接种试验、血清中和试验、酶联免疫吸附试验和乳胶凝集试验[7],其中PCR是最为灵敏、快速的诊断方法[8-9]。但是,在临床实际应用国标PCR方法进行临床疑似PR病例诊断时发现,直接用采集的鼻棉拭子、肺脏或扁桃体等样品处理后提取DNA,多数样本进行PCR得到的是阴性结果,但通过Vero细胞接种传代后,用细胞培养物提取DNA进行PCR得到的是阳性结果,推测可能是国标方法的灵敏度不够,样品中病毒含量较低时可能无法有效扩增。研究发现,套式PCR能够大幅度提高基因扩增的灵敏度,即使在模板浓度较低的情况下也能高效扩增目的基因[10]。为此,本研究旨在利用套式PCR技术对国标进行优化,提高检测灵敏度,以期在临床从组织病料中能够直接、有效地扩增目的片段,以达到快速、准确的诊断目的。
1材料与方法
1.1材料
1.1.1病毒PRV SXHX株为分离自户县某猪场野毒株;猪圆环病毒2型(PCV-2)SXXY13株分离自旬邑县某猪场;猪细小病毒(PPV)SXHZ株分离自汉中市某猪场;猪流行性腹泻病毒(PEDV)YLCH株分离自榆林某猪场;以上分离毒株均由咸阳职业技术学院畜牧兽医研究所动物疫病分子生物学诊断实验室保存。猪瘟病毒(CSFV)和猪蓝耳病病毒(PRRSV)均为普莱柯生物工程股份有限公司生产的疫苗毒株。
1.1.2病料采集或猪场送检的疑似PR病料(包括肺脏、脑组织和扁桃体)以及病猪血清,分别来自咸阳、宝鸡、渭南和西安地区猪场,共计8份,将病料研磨处理,12 000 r/min离心10 min,收集上清液置-70℃保存备用。
1.1.3细胞和试剂Vero细胞系为动物疫病分子生物学诊断实验室保存;Trizol Reagent、DNAzol Reagent为Invitrogen公司产品;AMV反转录酶(10 U/μL)、RNA酶抑制剂(40 U/μL)、DEPC处理水、rTaq酶(5 U/μL)、dNTPs(各成分均为10 mmol/L)、2×GC buffer等均购自宝生物工程(大连)有限公司。
1.1.4引物设计与合成参考国标GB/T 18641-2002,根据GenBank上公布的PRV gD基因序列(登录号AY196984、AY217094),设计并合成了2对引物,PRVgD-1F:5′- GCGGTCCCACGTTCGCATTCA -3′;PRVgD -1R:5′- CCCGGTCACGATTCGCAGCA-3;PRVgD-2F:5′-CACGGAGGACG- AGCTGGGGCT-3′ ;PRVgD-2R:5′- GTCCACGCCCCGCTTGAAGCT -3′。采用套式聚合酶链反应(nPCR)方法,预期扩增片段217bp,引物由生工生物工程(上海)有限公司合成,用DEPC处理水稀释到20 μmol/L。
1.2方法
1.2.1病毒DNA的提取取PRV SXHX株细胞培养物200 μL置于无菌EP管,加入DNAzol Reagent 1 000 μL,上下颠倒混匀,室温静置裂解10 min,4℃、12 000 r/min离心5 min,吸取600 μL上清液转移至另一无菌EP管,加入等体积冰冷无水乙醇沉淀10 min,4℃、12 000 r/min离心10 min,弃去上清液,加入1 mL冰冷的700 mL/L乙醇清洗2次,倒置干燥,用40 μL 8 mmol/L NaOH充分吹打溶解,置沸水浴中10 min,取出后冰浴3 min,在核酸蛋白测定仪上测定DNA的含量。
1.2.2PRV套式PCR(nPCR)方法的建立取已知浓度的DNA溶液作为模板,摸索扩增条件。第1次扩增反应体系中DNA 2.0 μL,2×GC buffer 12.5 μL,dNTPs 1.0 μL,PRVgD-1F、PRVgD -1R各0.5 μL,改变rTaqDNA聚合酶用量(0.25 μL~1.0 μL),用超纯水补足总体积25.0 μL。条件设定为:95℃ 5 min;94℃ 1 min,退火温度由60℃~65℃按1℃递增设定退火1 min,72℃ 1 min,共35个循环;最后72℃延伸10 min。第2次扩增时取2.0 μL第1次扩增产物作为模板,其他成分同第1次扩增进行设定摸索,只是引物更换为PRVgD -2F、PRVgD -2R。条件设定为:95℃ 5 min; 94℃ 1 min,退火温度由60℃~65℃按1℃递增设定退火1 min,72℃ 延伸1 min,共35个循环;最后72℃延伸10 min。确定反应体系和反应条件的最佳组合。
1.2.3PRV国标PCR方法与建立的nPCR方法灵敏性试验比较将DNA溶液做10倍梯度稀释至10-10倍,分别以各稀释后DNA溶液作为模板进行PCR扩增。国标法按照标准的操作要求进行。
建立的nPCR方法第1次扩增按照以下反应体系进行:DNA 2.0 μL,超纯水8.0 μL,2×GC buffer 12.5 μL,dNTP 1.0 μL,NPRVgD-1F、PRVgD -1R各0.5 μL,rTaq酶0.5 μL,总体积25.0 μL。PCR条件为:95℃ 5 min;94℃ 1 min,62℃ 1 min,72℃ 1 min,35个循环;最后72℃ 10 min。取第1次扩增产物2.0 μL作为模板进行第2次扩增,体系同第1次扩增,引物为PRVgD-2F/PRVgD-2R。PCR条件为95℃ 5 min;94℃ 1 min,65℃ 1 min,72℃ 1 min,共35个循环;72℃ 10 min。扩增完毕后取5.0 μL扩增产物,15 g/L琼脂糖凝胶中电泳,凝胶成像系统中观察照相。
1.2.4PRV nPCR方法特异性试验提取CSFV、PRRSV和PEDV等RNA病毒的核酸并反转录获得cDNA;按照DNAzol Reagent试剂说明提取PRV SXHX株、PCV-2 SXXY13株以及PPV SXHZ株等DNA病毒DNA模板,用建立的nPCR方法检测,PCR产物用15 g/L琼脂糖凝胶电泳,成像系统中观察照相。
1.2.5病料中PRV的检测对比用以上建立的检测方法和国标方法对收集的8份病料进行检测。
1.2.6病料中PRV的分离与检测分别取8份病料上清液200 μL,接种于Vero细胞盲传3代,出现细胞病变后反复冻融收集细胞液备用。提取8份盲传3代的Vero细胞液DNA,用国标PCR方法进行检测。
2结果
2.1PRV nPCR方法的建立
通过改变反应体系和反应条件,最终确定最优体系和条件为:第1次扩增DNA 2.0 μL,2×GC buffer 12.5 μL,超纯水8.0 μL,dNTP 1.0 μL,PRVgD-1F、PRVgD -1R各0.5 μL,rTaqDNA聚合酶0.5 μL,总体积25.0 μL。PCR条件为:95℃ 5 min;94℃ 1 min,62℃ 1 min,72℃ 1 min,35个循环;72℃延伸10 min。第2次扩增反应体系:取2.0 μL第1次扩增产物作为模板,体系其余组分同第1次扩增,引物为PRVgD-2F/PRVgD -2R。PCR条件为:95℃ 5 min;94℃ 1 min,65℃ 1 min,72℃ 1 min,35个循环;72℃延伸10 min。
2.2PRV国标PCR方法与建立的nPCR方法灵敏性试验结果
经过测定,PRV SXHX株细胞培养液DNA含量为260 ng/mL,将其按照10倍梯度进行稀释,分别进行PCR。从图1可知,国标法能够检测的极限为10-5倍稀释,即2.6×10-3ng/mL。所建立的nPCR方法能够扩增出10-8倍稀释的DNA(图2),即检测的极限为2.6×10-7ng/mL。表明建立的nPCR方法灵敏度提高了1 000倍。
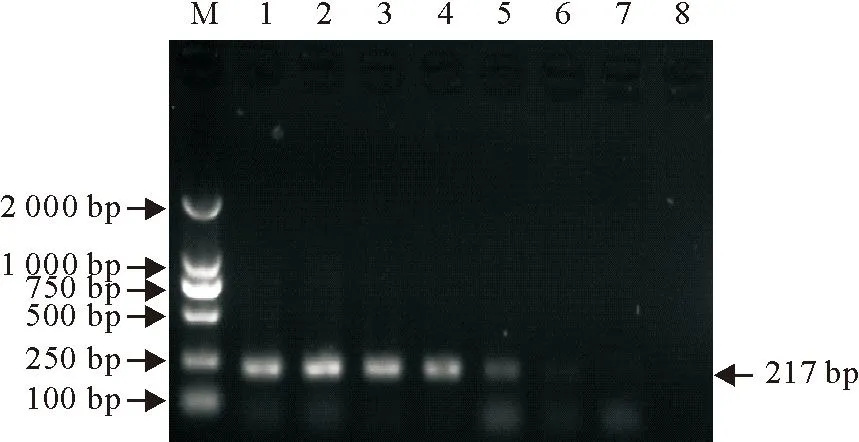
M. DNA标准DL 2 000;1~8. 依次为DNA浓度260×10-1ng/mL ~260×10-8ng/mL 10倍梯度稀释
M. DNA Marker DL 2 000; 1-8. 260×10-1ng/mL -260×10-8ng/mL of PRV DNA
图1PRV 国标PCR检测方法灵敏度试验结果
Fig.1The results of sensitivity test for PRV by PCR
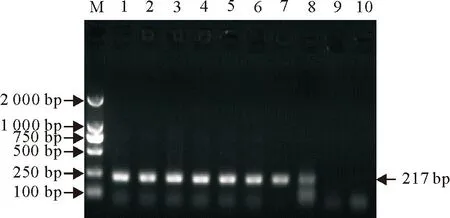
M. DNA标准DL 2 000;1~10.依次为DNA浓度 260×10-1ng/mL~260×10-10ng/mL 10倍梯度稀释
M. DNA Marker DL 2 000; 1-10. 260×10-1ng/mL -260×10-10ng/mL of PRV DNA
图2PRV nPCR检测方法灵敏度试验结果
Fig.2The results of sensitivity test for PRV by nPCR
2.3PRV nPCR检测方法特异性试验结果
用所建立的方法对CSFV、PRRSV、PRV SXHX株、PEDV、PCV-2 SXXY13株以及PPV SXHZ株cDNA/DNA进行扩增,结果发现,仅有PRV SXHX株扩增出了217 bp的预期目的条带,其他5种病毒均为阴性,提示所建立的方法特异性好(图3)。
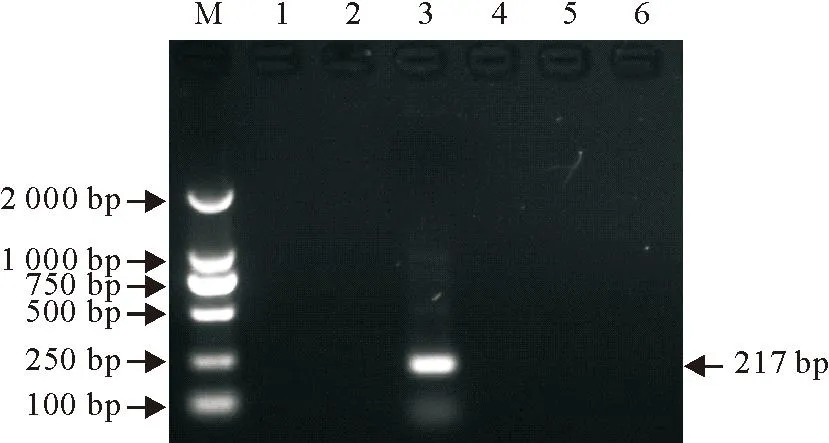
M.DNA标准DL 2 000;1.CSFV;2.PRRSV; 3.PRV; 4.PEDV;
5.PCV-2;6.PPV
M.DNA Marker DL 2 000; 1.CSFV;2.PRRSV; 3.PRV; 4.PEDV;
5.PCV-2;6.PPV
图3PRV nPCR检测方法特异性试验结果
Fig.3The result of specificity test for PRV by nPCR
2.4病料中PRV的检测结果对比
提取收集的8份疑似感染PRV的组织病料总DNA,分别用国标法和建立的nPCR方法进行检测,结果见图4。结果显示国标PCR方法对8份病料的扩增仅有2份出现目的条带,阳性率为25%;建立的nPCR方法在8份病料中5份扩增出了目的条带,阳性率为62.5%。结果显示,与国标法相比,优化后的检测方法能够更有效地从病料中扩增目的条带,大幅度提高了检出率。
2.5病料中PRV的分离与国标方法检测
经过Vero细胞3代盲传,采用国标方法从8份病料传代培养物中检测出了5份阳性结果,结果与改进后的nPCR方法完全一致。
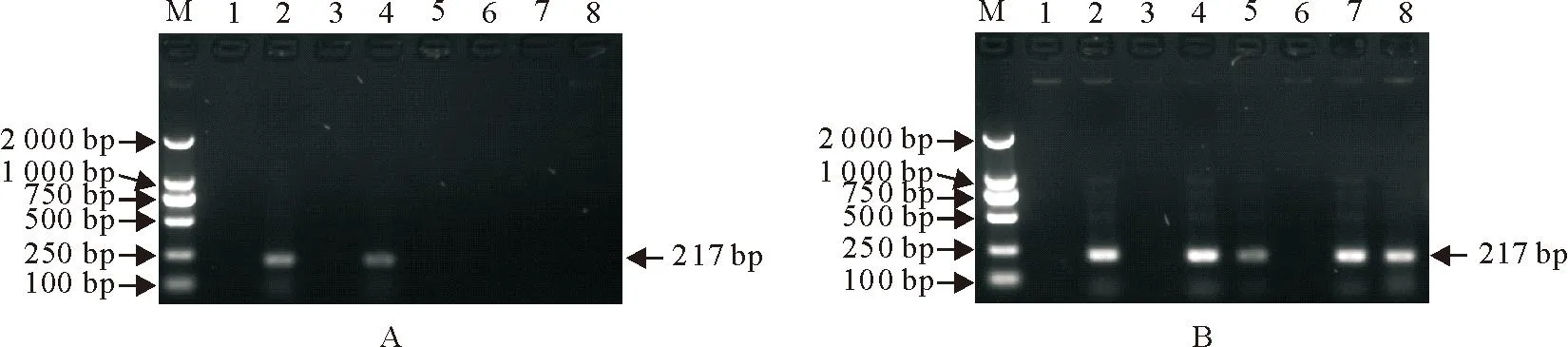
M. DNA标准DL 2 000; 1~8.病料检测
3讨论
猪伪狂犬病近年来在我国流行呈上升趋势,快速准确的诊断有助于对该病的有效防控。现在常规的诊断方法中,家兔接种试验耗时过长,常需要数天时间;病毒分离鉴定的优点是结果确实可靠,能获取毒株资源,但缺点是需要细胞培养,条件要求高,分离时间长;血清中和试验同样需要细胞培养;酶联免疫吸附试验优点是检测时间短,灵敏度高,但不足是只能反映抗体情况,不能确定病毒在机体内复制与分布情况。PCR技术具有灵敏度高、特异性好的优点,不需要细胞培养,所需时间短,能快速诊断,且诊断材料来源宽泛。国标中提供了检测PRV的PCR方法,但在实际应用中,笔者发现对于PR临床疑似病例国标方法一些检测阴性材料通过细胞接种后检测呈阳性,提示国标方法灵敏度较低,存在漏检情况。本研究根据套式PCR原理,在国标gD基因引物扩增片段之外再设计一对引物,建立了检测PRV的套式PCR方法。
引物的设计是PCR检测技术的关键,引物决定了检测的灵敏度和特异性[11-12]。本研究采用设计的外扩引物建立的nPCR方法,经过对CSFV、PRRSV、PEDV、PCV-2以及PPV cDNA/DNA扩增,结果均为阴性,仅能从PRV参考毒株中获得阳性结果,证实设计的引物是特异、可靠的。PCR扩增条件的优化是获得良好结果的保证,由于PRV DNA G+C含量高达73%,变性时解链难度大,因此,在提取DNA后先沸水浴10 min,然后迅速置于冰水中冷却,目的就是获得尽量多的单链DNA。在反应体系中,笔者在试验中参考文献曾尝试了DMSO、甘油等成分[13],均未取得理想结果,推测可能与引物设计不同有关,但使用2×GC buffer后能稳定获得目的条带,是成功扩增PRV基因的关键成分。灵敏度试验发现,建立的nPCR方法能够检测的极限为2.6×10-7ng/mL,比国标方法灵敏度提高了1 000倍,对临床病毒含量低的病料检测有重要意义,通过对8份疑似病料的检测也证实了该方法确能大幅度的提高检出率,为临床检测提供了一种可靠的检测手段。
参考文献:
[1]殷震,刘景华.动物病毒学[M].第2版.北京:科技出版社,1997.
[2]陈焕春,方六荣,何启盖,等.猪伪狂犬病病毒鄂A株的分离鉴定[J].畜牧兽医学报,1998,29(2):97-104.
[3]程晶,荫硕焱,盖新娜,等.2006-2008年我国部分地区规模化猪场PRV血清流行病学调查[J].中国动物传染病学报,2009,17(1):67-71.
[4]彭金美,安同庆,赵鸿远,等.猪伪狂犬病病毒新流行株的分离鉴定及抗原差异性分析[J].中国预防兽医学报,2013,35(1) :1-4.
[5]吕素芳,郭广君,魏凤,等.猪伪狂犬病病毒gE-/gI-/TK-多基因缺失活疫苗对猪的安全性与免疫效力研究[J].动物医学进展,2014,35(4):1-5.
[6]张显浩,陈瑞爱,李冰,等.2012-2013年我国集约化猪场猪伪狂犬病病毒感染情况的调查[J].动物医学进展,2015,36(3):133-136.
[7]陈焕春,何启盖,李晓成,等.GB/T 18641-2002伪狂犬病诊断技术[S].北京:中国标准出版社,2002.
[8]Sami L,Ursu K,McKillen J,et al.Simultaneous detection of three porcine viruses by multiplex PCR[J].Acta Vet Hung,2007,55 (2):267-276.
[9]焦明,赵玉军,庄金秋,等.猪伪狂犬病诊断方法研究进展[J].动物医学进展,2007,28(1):88-91.
[10]吴旭锦,朱小甫.新城疫国标RT-PCR诊断方法的优化与应用[J].西北农林科技大学学报:自然科学版,2015,43(3):27-31.
[11]娄高明,杜伟贤,廖筱萍,等.PCR检测伪狂犬病病毒DNA[J].中国生物化学与分子生物学报,2001,1(4):519- 523.
[12]余丽芸,刘建柱,侯喜林,等.多重PCR检测猪瘟病毒、猪细小病毒和猪伪狂犬病病毒的研究—猪瘟病毒、猪细小病毒和猪伪狂犬病病毒多重PCR引物设计[J].动物医学进展,2004,25(6):75-77.
[13]徐葵,邱志明.DMSO对PCR扩增反应的影响[J].昆明医学院学报,2001,(1) :77-79.
Establishment and Application of nPCR for Detecting Porcine Pseudorabies Virus
WU Xu-jin ,ZHU Xiao-fu
(AnimalEpidemicDiseaseDiagnosticLaboratoryofMolecularBiology,InstituteofAnimalHusbandryandVeterinaryMedicine,XianyangVocationalTechnicalCollege,Xianyang,Shaanxi,712000,China)
Abstract:To establish a highly sensitive nested PCR methods for detecting porcine pseudorabies virus,according to published gene sequences of PRV gD, two pairs of primers were designed and synthesized, the nested PCR method was established by optimizing the reaction system and conditions. The method applicability was verified by sensitivity test, specificity test, sample comparison tests. The results showed that the detection limit of the method established is 2.6 × 10-7ng/mL, the sensitivity is 1 000 times higher than that of national standard method, and the objective band was only amplified from the pseudorabies virus reference strain, and could greatly improve the positive detection rate from suspected samples. The nested PCR detection mehod with high sensitivity and good specificity for rapid PRV detection was successfully established.
Key words:Pseudorabies virus; nested PCR; diagnosis; application
收稿日期:2015-12-15
基金项目:咸阳市科学技术研究计划项目(2015k03-21)
作者简介:吴旭锦(1979-),女,博士,副教授,主要从事动物疫病分子病原学与纳米药物学研究。*通讯作者
中图分类号:S852.65
文献标识码:A
文章编号:1007-5038(2016)06-0018-04
猜你喜欢
体育时空(2016年9期)2016-11-10
科技视界(2016年21期)2016-10-17
中国实用医药(2016年24期)2016-10-17
科学与财富(2016年28期)2016-10-14
考试周刊(2016年76期)2016-10-09
科技视界(2016年20期)2016-09-29
科技视界(2016年20期)2016-09-29
科技视界(2016年20期)2016-09-29
科技视界(2016年20期)2016-09-29
大众理财顾问(2016年8期)2016-09-28
