
瑞加德松的全合成研究
2016-03-23 23:36:37刘伟罗剑飞张志刚
上海医药 2016年3期
刘伟+罗剑飞+张志刚
摘 要 目的:改进瑞加德松的合成工艺,提高收率与质量,获得良好的生产工艺。方法:以2-氯腺苷的羟基为原料,经叔丁基二甲基硅烷保护,与水合肼反应得2, 3, 5-三叔丁基二甲基硅氧基-2-肼基腺苷,再和2-甲酰基-3-氧代丙酸乙酯环合后经甲胺化得带保护的瑞加德松,最后脱去保护基得到瑞加德松。结果:产品纯度和收率分别提高到99.0%和47.2%。结论:优化后工艺收率高,产品质量好,操作简便,适合工业化生产。
关键词 瑞加德松 合成 工艺改进 纯化
中图分类号:O626.21 文献标识码:A 文章编号:1006-1533(2016)03-0073-03
Total synthesis of regadenoson
LIU Wei*, LUO Jianfei, ZHANG Zhigang
(Shanghai Ziyuan Pharmaceutical Co. Ltd., Shanghai 201108, China)
ABSTRACT Objective: To improve the process for the synthesis of regadenoson and its quality. Methods: Regadenoson
was synthesized from 2-chloroadenosine via etherification with t-butyldimethylchlorosilane, reaction with hydrazine hydrate to give 2,3,5-tri-(tert-butyldimethylsiyl)oxy-2-hydrazinyl adenosine, which was cyclized with ethyl 2-formyl-3-oxopropanoate, and finally the protecting groups were removed. Results: The purity and yield of regadenoson were improved up to 99.0% and 47.2%, respectively. Conclusion: The improved process can be easily operated with high yield and quality, and is suitable to industrial production.
KEY WORDS regadenoson; synthesis; process improvement; purification
瑞加德松(regadenoson,1)是一种具有高度选择性的腺苷A2A受体激动剂,可通过激活A2A腺苷受体,促进冠脉舒张和增加冠脉血流量,在心肌灌注造影研究中显示其应激安全、有效[1-2]。本品由CV Therapeutics公司开发,2008年4月美国FDA批准上市,用于心肌灌注显像。
文献报道的合成路线主要有以下两条:①以2-氯腺苷为底物,与水合肼反应,生成2-肼基腺苷,然后与2-甲酰基-3-氧代丙酸乙酯环合生成1-(6-氨基-9-β-D-呋喃核糖基-9H-嘌呤-2-基)-1H-吡唑-4-甲酸乙酯,该化合物在50~70 ℃下在压力反应器中与甲胺水溶液反应,生成化合物1[3-4],总收率约43%,但需要在加热条件下于压力反应器中反应,对设备要求较高,导致成本升高,安全系数降低,不利于放大生产。②以1H-吡唑-4-羧酸为原料,酯化所得1H-吡唑-4-羧酸甲酯与甲胺反应得N-甲基-1H-吡唑-4-甲酰胺,再与2-碘腺苷在强碱作用下得1[5-6],此法路线较短,总收率约36%,但需用昂贵的2-碘腺苷。本文参考路线①的方法,针对上述问题加以研究与改进,优化了反应及纯化等条件,提高了产品收率和质量,使整个合成工艺能够满足工业化生产的要求。
1 仪器与试药
400 MR核磁共振仪(美国Agilent公司);RE-52C旋转蒸发仪(巩义市予华仪器有限公司);JH50熔点仪(上海佳航仪器仪表有限公司);电子天平(上海伊准电子科技有限公司)。
2-氯腺苷(纯度99%)、叔丁基二甲基氯化硅、2-甲酰基-3-氧代丙酸乙酯和四丁基氟化铵(迈瑞尔化学有限公司);水合肼、甲胺、N, N-二甲基甲酰胺、二氯甲烷、四氢呋喃、乙腈和无水乙醇(国药集团化学试剂有限公司);其他试剂均为市售分析纯。
2 方法
以国内已有批量供应的2-氯腺苷为起始原料,经过5步反应合成目标产物瑞加德松(图1)。
2.1 2, 3, 5-三叔丁基二甲基硅氧基-2-氯腺苷(2)的制备
将2-氯腺苷((50.0 g,0.17 mol)溶于500 ml吡啶中,搅拌下加入叔丁基二甲基氯硅烷(210 g,1.40 mol)和咪唑(90.0 g,1.32 mol),室温反应24 h。减压浓缩除去吡啶,加入500 ml二氯甲烷使剩余固体溶解。然后,先用饱和碳酸氢钠溶液洗涤两次,再用饱和氯化钠溶液洗涤三次,二氯甲烷相用无水硫酸钠干燥,过滤,减压浓缩,得化合物2(91.8 g,收率86%),纯度92%;ESI-MS(m/z):666[M+Na]+;1H NMR (400 MHz,DMSO-d6) δ 8.32 (s,1H),7.82 (s,2H),5.80 (d,J=6.1 Hz,1H),4.95–4.79 (m,1H),4.31 (s,1H),3.97 (s,2H),3.70 (dd,J=13.2,6.7 Hz,1H),0.87(d,J=13.6 Hz,18H),0.71 (s,9H),0.12–0.02 (m,12H),-0.13 (s,3H),-0.35(s,3H)。
2.2 2, 3, 5-三叔丁基二甲基硅氧基-2-肼基腺苷(3)的制备[7]
将化合物2(50.0 g,0.078 mol)溶解于200 ml无水乙醇中,50 ℃下搅拌溶解。氮气保护下,加入80%一水合肼(80.0 g,1.28 mol),温度升至70 ℃反应20 h。将反应液缓慢倒入2 L冰水中,搅拌1 h,过滤。滤饼用纯化水洗涤三次,过滤,干燥得化合物3(46.7 g,收率94.0%),纯度92%;ESI-MS(m/z):662[M+Na]+;1H NMR (400 MHz,DMSO-d6) δ 7.92 (s,1H),7.28 (s,1H),6.83 (s,2H),5.78 (d,J=6.3 Hz,1H),4.87–4.63(m,1H),4.22 (s,1H),4.03–3.81 (m,4H),3.71(d, J=8.0 Hz,1H),0.88 (d,J=5.0 Hz,18H),0.72 (s,9H),0.06 (d,J=8.8 Hz,12H),-0.11 (s,3H),-0.31 (s,3H)。
2.3 1-(6-氨基-9-β-D-呋喃核糖基-9H-嘌呤-2-基)-1H-吡唑-4-甲酸乙酯的叔丁基二甲基硅基保护衍生物(4)的制备[8]
在氮气保护下将化合物3(50.0 g,0.078 mol)溶解于500 ml异丙醇中,加热至80 ℃,加入2-甲酰基-3-氧代丙酸乙酯(13.0 g,0.090 mol),反应2 h。将反应液倒入4 L冰冷的纯化水中,搅拌30 min,过滤,以冰冷的异丙醇-水(1∶6)的混合液洗涤滤饼2次,每次100 ml,滤饼经55 ℃真空干燥得化合物4(52.6 g,收率90.0%),纯度88%;ESI-MS(m/z):770[M+Na]+;1H NMR (400 MHz,DMSO-d6) δ 8.86 (s, 1H), 8.37 (s, 1H),8.04 (s, 1H), 7.83 (s, 2H), 5.90 (d, J = 5.7 Hz, 1H), 4.98(s, 1H), 4.32 (s, 1H), 4.24 (q, J = 7.0 Hz, 2H), 4.08 – 3.91(m, 2H), 3.72 (d, J = 7.7 Hz, 1H), 1.27 (t, J = 7.0 Hz,4H), 0.87 (d, J = 18.6 Hz, 18H), 0.70 (s, 9H), 0.08 (dd, J= 20.6, 9.9 Hz, 12H), -0.12 (s, 3H), -0.32 (s, 3H)。
2.4 1-(6-氨基-9-β-D-呋喃核糖基-9H-嘌呤-2-基)-N-甲基-1H-吡唑-4-甲酰胺(1)的制备[9]
在常压下,将化合物4(25.0 g,0.033 mol)溶解于500 g含甲胺的甲醇溶液(40%)中,升温至65 ℃,反应24 h。30 ℃减压除去甲胺,常温下,向反应液中直接加入四丁基氟化铵(25.0 g,0.096 mol),反应12 h。固体析出,过滤,滤饼加入乙醇-水(1∶5)的混合液200 ml,搅拌2 h,过滤,滤饼用30 ml纯化水洗涤,于55 ℃减压烘干,得白色固体(7.16 g,收率64.9%),mp 179~186 ℃。纯度99.0%(图2) [HPLC法:色谱柱Agilent XDB-C18柱(4.6 mm×150 mm,10 μm);流动相A:水;B:乙腈,梯度洗脱(0→5 min: A95%;5→30 min: A95%→90%;30→50 min: A90%→50%);检测波长254 nm;柱温25 ℃;流速1.0 ml/min]。ESIMS(m/z):413[M+Na]+;1HNMR(400 MHz,CD3OD)δ 9.05 (s,1H),8.38 (s,1H),7.11 (s,1H),6.06 (d,J=6.4 Hz,1H),4.64 (m,1H),4.34 (d,J=6.4 Hz,1H),4.13 (d,J=4.8 Hz,1H),3.91 (m,1H),3.78 (m,1H),2.90 (s,3H);13C NMR (100 MHz,DMSO-d6) δ 162.1,156.8,151.0,150.7,141.3,140.4,130.0,120.6,118.3,87.3,86.2,74.1,71.0,61.9,26.0。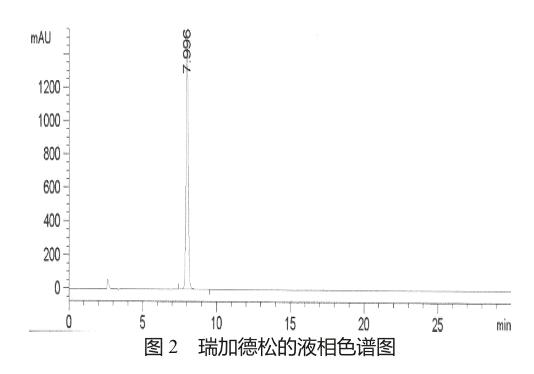
3 结果
按上述步骤多次试验,结果表明本工艺操作简便,反应容易控制,产品收率与质量稳定,产品纯度经HPLC检测达到了99.0%,产品总收率以2-氯腺苷计达到了47.2%,高于文献报道路线①的43%,产品经NMR,MS,熔点等分析确证结构。
4 讨论
第1步采用叔丁基二甲基硅基作为2-氯腺苷的糖苷保护基,提高了中间体的稳定性,使得后续反应的转化率提高,过程杂质更容易得到控制。反应结束后只需用饱和碳酸氢钠和饱和氯化钠溶液萃洗即可得到纯度较高中间体。
第4步为胺酯交换反应,文献中使用了加压设备,本实验通过改换反应溶剂和温度,使得该反应在常压反应容器中能够顺利进行,降低了对反应设备的要求,操作更加简便,更适合放大生产。反应结束后经过简单处理,不需纯化即可进行下一步反应。
参考文献
[1] Al Jaroudi W, Iskandrian AE. Regadenoson: a new myocardial stress agent[J]. J Am Coll Cardiol, 2009, 54 (13): 1123 - 1130.
[2] Lieu HD, Shryock JC, von Mering GO, et al. Regadenoson, a selective A2A adenosine receptor agonist, causes dose dependent increases in coronary blood flow velocity in humans [J]. J Nucl Cardiol, 2007, 14(4): 514-520.
[3] Zablocki J, Palle V, Blackburn B, et al. 2-Substituted pi system derivatives of adenosine that are coronary vasodilators acting via the A2A adenosine receptor [J]. Nucleosides Nucleotides Nucleic Acids, 2001, 20(4-7): 343-360.
[4] Grisenti P, Shahrzad PE, Guazzi G, et al. Stable solid forms of regadenoson: WO2014167046A1[P]. 2014-10-16.
[5] Linden JM, Sullivan GW, Scheld WM. Method compositions for treating inflammatory response: US6514949[P]. 2003-02-04.
[6] Ma C, Nadji S, Wooldridge LT. Improved processes for the preparation of regadenoson and a new crystalline form thereof: WO2012149196A1[P]. 2012-11-02.
[7] Montgomery JA, Holum LB. Synthesis of potential anticancer agents. Ⅲ. Hydrazino analogs of biologically active purines[J]. J Am Chem Soc, 1957, 79(9): 2185-2188.
[8] Lieu H, Blackburn B, Belardinelli L. Methods and compositions for increasing patient tolerability during myocardial imaging methods: US20090081120A1[P]. 2009-03-26.
[9] Kvapil L, Hradil P, Grepl M, et al. Polymorph of 2-[4-[(methylamino) carbonyl]-1H-pyrazol-1-yl] adenosine: US20140323712A1[P]. 2014-10-30.
猜你喜欢
湖北农业科学(2016年20期)2017-02-15 17:52:38
考试周刊(2016年85期)2016-11-11 02:09:06
农业与技术(2016年15期)2016-11-09 07:10:15
科技视界(2016年18期)2016-11-03 00:34:31
中国科技博览(2016年16期)2016-09-19 00:34:50
中国科技博览(2016年10期)2016-04-29 09:52:53
中国科技博览(2016年10期)2016-04-29 04:50:02
中国科技博览(2016年6期)2016-04-25 15:46:56
科技视界(2015年30期)2015-10-22 10:13:55
科技资讯(2015年20期)2015-10-15 19:47:25
