
小麦TaGAPC定点突变体基因载体构建及其原核表达
2016-03-18 08:39:19郭子平翟清华曾令锋邓西平杨淑慎
华北农学报 2016年1期
关键词:原核表达
郭子平,翟清华,曾令锋,邓西平,杨淑慎
(1.西北农林科技大学 生命科学学院,陕西 杨凌 712100;2.西北农林科技大学 黄土高原土壤侵蚀与旱地农业国家重点实验室,陕西 杨凌 712100)
小麦TaGAPC定点突变体基因载体构建及其原核表达
郭子平1,翟清华1,曾令锋1,邓西平2,杨淑慎1
(1.西北农林科技大学 生命科学学院,陕西 杨凌712100;2.西北农林科技大学 黄土高原土壤侵蚀与旱地农业国家重点实验室,陕西 杨凌712100)
摘要:为了研究催化活性半胱氨酸Cys154、Cys158残基对小麦细胞质甘油醛-3-磷酸脱氢酶(TaGAPC)蛋白酶活性的影响,利用重叠延伸PCR法分别将这2个位点的半胱氨酸突变成丝氨酸,并分别连入pET28a(+)原核表达载体。在25 ℃条件下,TaGAPC及其定点基因TaCys154S/TaCys158S经0.25 mmol/L IPTG诱导后高效表达,SDS-PAGE电泳检测结果显示,目标重组蛋白均为可溶性且大小约为40 kDa,与预测结果一致。在此基础上,经超声波破菌及镍柱纯化获得3种纯化重组蛋白。这为后续TaGAPC及其定点突变体蛋白酶活性测定及活性位点分析提供试验材料。
关键词:TaGAPC;重叠延伸PCR;定点突变;原核表达;蛋白纯化
甘油醛-3-磷酸脱氢酶(Glyceraldehyde-3-phosphate dehydrogenase,GAPDH)作为糖酵解反应的一种关键酶,在烟酰胺腺嘌呤二核苷酸(辅酶Ⅰ)和无机磷酸盐存在的情况下,催化3-磷酸-甘油醛氧化生成1,3-二磷酸甘油醛[1]。GAPDH普遍存在于原核和真核生物中,具有高度种属保守序列[2-3]。长期以来,GAPDH由于在同种组织或细胞中相对恒定的表达量,所以一直被当作“管家基因”和内参[4]。然而近年来大量研究发现,GAPDH除参与糖酵解反应外,还参与许多其他代谢过程,是一种多功能蛋白质。在哺乳动物细胞中,GAPDH作为一种介导细胞质和细胞核之间信息传递的穿梭蛋白,其广泛的生理功能涉及DNA复制与修复、基因转录、细胞凋亡、前体tRNA的运输、组蛋白基因的表达调控、端粒结构调节、核膜融合和微管成束等[5-9]。然而有关植物GAPDH参与非糖酵解途径的功能研究则不甚清楚。
在高等动植物中,局部序列比对显示GAPDH蛋白底物结合位点区的第150-161的氨基酸序列高度保守,而位于第154位(Cys154)、第158位(Cys158)的半胱氨酸Cys(相应在拟南芥中的编号分别为Cys149和Cys153)是GAPDH蛋白酶催化活性位点[10]。GAPDH蛋白活性位点的Cys残基巯基易受不同种类的翻译后氧化还原修饰(PTMs)以响应不同的ROS-和RNS-依赖的氧化还原信号[11]。GAPDH蛋白的PTMs包括磺酸化、亚硝基化和谷胱甘肽化等,这些修饰不但影响GAPDH酶活性导致其部分或全部失活,并且进一步影响GAPDH与其他蛋白的相互作用,导致GAPDH蛋白定位改变,引发其多样性功能[10,12-13]。在拟南芥中,细胞质AtGAPC (AtGAPC1和AtGAPC2)活性位点半胱氨酸残基巯基S-亚硝基化导致蛋白可逆失活[14-15],而AtGAPC1酶活性严格依赖催化Cys149,并且该残基对H2O2极为敏感,但定点突变Cys153对酶活性几乎无影响[16]。H2O2通过修饰活性位点的Cys残基巯基促进AtGAPC与磷脂酶Dδ(Phospholipase Dδ,PLDδ)的相互作用,进而介导响应ABA和水分胁迫[17]。在粟酒裂殖酵母 (Schizosaccharomycespombe)中,GAPDH的Cys残基被H2O2瞬时氧化后促进GAPDH与MAPK途径中Mcs4相互作用,但是点突变Cys152造成该相互作用减弱,进而影响H2O2胁迫信号传导[18]。Piattoni等[19]推测小麦细胞质TaGAPC蛋白活性位点的Cys154可能至少是受H2O2等氧化作用的一个重要氨基酸,但并未有后续试验对该推测及另外一个位点Cys158对酶活性的影响进行证实和研究。
本试验在已构建成功的pMD19T-TaGAPC载体的基础上,通过重叠延伸PCR技术分别获得TaGAPC保守区活性位点Cys154、Cys158定点突变基因全长TaCys154S、TaCys158S,然后构建原核表达载体,在大肠杆菌中用IPTG诱导表达得到可溶性的重组蛋白并进行了纯化,这为后续TaGAPC及其定点突变体蛋白酶活性测定及TaGAPC催化活性位点的分析提供论证基础。
1材料和方法
1.1试剂和载体及菌种
高保真DNA聚合酶pfu、DNA聚合酶Taq、T4DNA 连接酶、IPTG(异丙基-β-D-硫代吡喃半乳糖苷)、X-gal(5-溴-4-氯-3-吲哚-β-D半乳糖苷)、Tris(三羟甲基氨基甲烷)、克隆载体pGEM-T easy、DNA琼脂糖凝胶回收试剂盒均购自TIANGEN公司;限制性核酸内切酶NdeⅠ和BamHⅠ、DNA分子量标记DL15000和DL2000购自THERMO公司;Ni-Agrose His标签蛋白纯化试剂盒(可溶性蛋白)购自CWBIO公司;大肠杆菌DH5α菌株和pMD19T载体为西北农林科技大学生命科学学院杨淑慎教授实验室保存。pET28a(+)原核表达质粒由西北农林科技大学生命科学学院陈坤明教授惠赠。
1.2引物
所有引物均合成于北京奥科鼎盛生物科技有限公司,其序列如下(表1)。

表1 本试验所用引物
注:斜体部分为保护碱基;下划线部分为酶切位点;粗体部分为引入的定点突变碱基。
Note:Italics sections are protective bases;Underlined sequences are restriction enzyme sites;Bold sequences are the site-directed bases.
1.3试验方法
1.3.1pET28a(+)-TaGAPC表达载体的构建以野生型靶基因TaGAPC(NCBI登录号:N° EF592180,基因全长1 014 bp,已克隆在pMD19T载体上,重组质粒命名为pMD19T-TaGAPC)为模板,设计引物TaGAPC-for、TaGAPC-rev扩增TaGAPC基因,PCR扩增体系为20 μL:TaqBuffer 4 μL,MgCl21.5 μL,dNTP 0.8 μL,正、反向引物各0.5 μL,模板1 μL,Taq聚合酶0.2 μL,ddH2O 11.5 μL;PCR反应体系为:94 ℃预变性3 min;94 ℃变性30 s,58 ℃退火45 s,72 ℃延伸1 min,32个循环;72 ℃延伸7 min。反应完成后在1%琼脂糖凝胶电泳上检测PCR产物,DNA胶回收试剂盒纯化回收位置正确的条带。之后对纯化的PCR产物、pET28a(+)质粒进行NdeⅠ、BamHⅠ双酶切并连接。连接反应体系为10 μL:TaGAPC片段6 μL、pET28a(+)载体2.0 μL、T4Buffer 1 μL、T4连接酶1 μL轻柔混匀后16 ℃连接过夜。取连接产物转化大肠杆菌DH5α,然后涂布于LB平板上(含100 mg/mL卡那霉素)培养至长出单菌落,挑取单菌落进行PCR检测和提取重组质粒进行双酶切验证,并送交北京奥科公司测序验证。
1.3.2pGEM-T easy-TaCys154S/TaCys158S载体的构建利用重叠延伸PCR法将TaGAPC基因第154,158位的半胱氨酸残基突变成丝氨酸[20-22]。以TaGAPC基因为模板,采用pfu聚合酶,在第一轮PCR中,以TaGAPC-for/TaCys154S-down、TaCys154S-up/TaGAPC-rev,或TaGAPC-for/TaCys158S-down、TaCys158S-up/TaGAPC-rev这4对引物分别扩增TaCys154S/TaCys158S基因小片段。PCR反应体系为50 μL:10×pfu Buffer(+MgSO4) 5 μL,dNTP 5 μL,正、反向引物各1 μL,模板1 μL,pfu聚合酶1 μL,ddH2O 36 μL。反应参数为:94 ℃预变性3 min;94 ℃变性30 s,60 ℃退火45 s,72 ℃延伸40 s,32个循环;72 ℃延伸7 min。反应完成后在1%琼脂糖凝胶电泳上检测PCR产物,DNA胶回收试剂盒纯化回收位置正确的条带。
以上述分别获得的TaCys154S/TaCys158基因小片段为模板,进行第二轮PCR反应合成相应的全长片段。PCR反应体系为20 μL:10×pfu Buffer(+MgSO4) 5 μL,dNTP 1 μL,TaGAPC-for、TaGAPC-rev引物各0.5 μL,模板分别为2 μL/1 μL,pfu高保真聚合酶0.2 μL,ddH2O 9.8 μL。反应参数为:94 ℃预变性3 min;94 ℃变性30 s,60 ℃退火45 s,72 ℃延伸1 min,32个循环;72 ℃延伸7 min。反应完成后电泳检测PCR产物,取条带位置正确的PCR产物进行末端加尾反应,体系为100 μL:4 μL dNTP,0.5 μLTaq聚合酶,95.5 μL PCR产物,于72 ℃,PCR延伸15 min,反应完成后DNA胶回收试剂盒纯化回收产物。
将TaCys154S/TaCys158S基因全长片段分别连接至pGEM-T easy载体。将基因全长片段5.7 μL、pGEM-T easy载体0.3 μL、T4Buffer 3 μL、T4连接酶1 μL共10 μL轻柔混匀后16 ℃连接过夜。取连接产物转化大肠杆菌DH5α,然后涂布于LB平板上(含100 mg/mL卡那霉素,24 mg/mL IPTG,及20 mg/mL X-gal),37 ℃倒置培养。挑取单克隆进行菌落PCR检测和提取质粒进行双酶切验证,将阳性质粒送交北京奥科公司测序。
1.3.3pET28a(+)-TaCys154S/TaCys158S原核表达载体的构建对重组质粒pGEM-T easy-TaCys154S/TaCys158S进行NdeⅠ、BamHⅠ双酶切试验。双酶切体系为50 μL:10×Tango Buffer 5 μL,NdeⅠ0.5 μL,BamHⅠ0.5 μL,质粒20 μL,ddH2O 24 μL。酶切反应于37 ℃水浴中保持12 h后,利用DNA胶回收试剂盒纯化回收位置正确的条带。然后将其连接到同样经NdeⅠ、BamHⅠ双酶切的pET28a(+)载体。连接反应体系为10 μL:TaCys154S/TaCys158S基因全长片段6 μL、pET28a(+)载体2.0 μL、T4Buffer 1 μL、T4连接酶1 μL轻柔混匀后16 ℃连接过夜。取连接产物转化大肠杆菌DH5α,然后涂布于LB平板上(含100 mg/mL卡那霉素),37 ℃倒置培养。挑取单克隆进行菌落PCR检测和提取质粒进行双酶切验证,将阳性质粒送交北京奥科公司测序。
1.3.4TaGAPC和TaCys154S/TaCys158S基因的原核表达纯化将已构建的原核表达载体pET28a(+)-TaGAPC/TaCys154S/TaCys158S分别转化大肠杆菌表达菌株BL21(DE3)进行诱导表达,方法参照[16,23],但略有优化。在37 ℃培养的LB平板(含100 mg/mL卡那霉素)上挑取阳性单菌落进行扩大培养,至OD600为0.4~0.6,于25 ℃,0.25 mmol/L IPTG诱导表达12 h,6 500 r/min离心收集菌体,25 mmol/L Tris-HCl(pH值7.5)清洗2次后放于-80 ℃保存。超声波工作条件为:250 W,工作4 s,间歇6 s,共1 h。SDS-PAGE电泳检测表达蛋白均为可溶性蛋白,10 400 r/min离心2次取上清,使用Ni-Agrose His标签蛋白纯化试剂盒(可溶性蛋白)纯化蛋白,按照说明书中的步骤操作。SDS-PAGE电泳检测蛋白纯化效果。
2结果与分析
2.1pGEM-T-easy-TaCys154S/TaCys158S克隆重组载体的构建
通过重叠延伸PCR法在TaGAPC基因中引入定点突变,即将Cys154、Cys158残基定点突变成丝氨酸,之后连接pGEMT-easy载体,转化大肠杆菌并培养单克隆。对菌落PCR检测为阳性的单克隆进行测序,结果显示定点突变成功。重组质粒经NdeⅠ、BamH Ⅰ双酶切的电泳检测结果如图1所示。
2.2 pET28a(+)-TaGAPC/TaCys154S/TaCys158S原核表达重组载体的构建
对扩增的TaGAPC产物及重组质粒pGEMT-easy-TaCys154S/TaCys158S,分别经NdeⅠ、BamHⅠ双酶切后电泳检测,并分别纯化回收目的条带,将其连接到经同样NdeⅠ、BamHⅠ双酶切的pET28a(+)表达载体。选取菌落PCR检测为阳性的单菌落,摇菌提取质粒进行双酶切及测序验证,得到约1 014 bp片段(图2),与预期大小一致,说明重组原核表达载体pET28a(+)-TaGAPC/TaCys154S/TaCys158S构建成功。

M.DL15000分子量标准;1.pGEM-T easy-TaCys154S
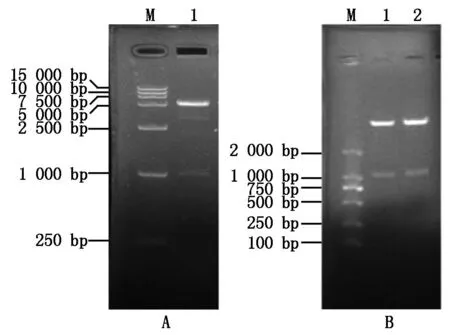
A:M.DL15000分子量标准;1.pET28a(+)-TaGAPC双酶切片段;B:M.DL2000分子量标准;1.pET28a(+)-TaCys154S双酶切片段;2.pET28a(+)-TaCys158S双酶切片段。
A:M.DL15000 DNA Marker;1.Restriction enzyme fragments of pET28a(+)-TaGAPC;B:M.DL2000 DNA Marker;1.Restriction enzyme fragments of pET28a(+)-TaCys154S;2.Restriction enzyme fragments of pET28a(+)-TaCys158S.
图2重组质粒pET28a(+)-TaGAPC(A)和
pET28a(+)-TaCys154S/TaCys158S(B)酶切鉴定
Fig.2Restriction enzyme digestion of the recombinant
expression vectors pET28a(+)-TaGAPC(A) and
pET28a(+)-TaCys154S/TaCys158S(B)
2.3TaGAPC和TaCys154S/TaCys158S重组蛋白的原核表达及纯化
将TaGAPC及其TaCys154S/TaCys158S原核表达重组质粒分别转化E.coliBL21(DE3),于37 ℃条件下,200 r/min摇菌至OD600为0.4~0.6,在25 ℃条件下,0.25 mmol/L ITPG诱导表达12 h,离心收集菌体,经超声波破菌及Ni-IDA柱纯化得到可溶性重组蛋白(图3)。从图3可看出,纯化蛋白分子量约为40.0 kDa(纯化蛋白N端含6×His标签),与预期结果一致,并且获得较高纯度的重组蛋白。
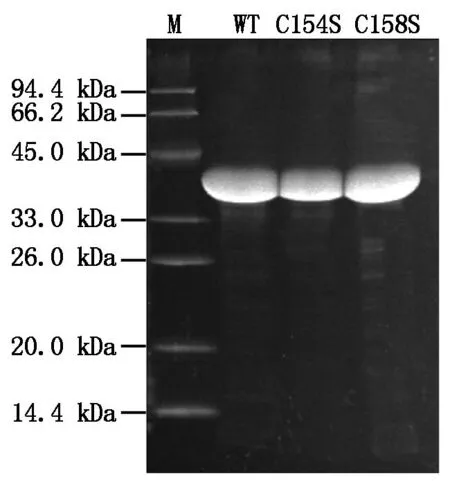
M.蛋白Marker(LOW);WT.TaGAPC蛋白;
3讨论
作为糖酵解代谢的一种关键酶,GAPDH催化3-磷酸甘油醛氧化为生命活动提供能量[1]。GAPDH基因几乎在各组织或器官中相对稳定且高水平表达,故被广泛用作标准化内参研究生物体内其他基因和蛋白的表达[4,24]。但最近大量研究表明,GAPDH 是一种多功能蛋白质,除参与糖酵解以外,还参与其他非生物代谢过程[5-9]。相对于动物细胞仅含有一种GAPDH同工酶,高等植物GAPDH包含4种由不同基因编码的同工酶,不同的同工酶由于其亚细胞定位不同导致其参与的生理过程具有多样性[7]。
在高等植物中,位于GAPDH底物结合区约150-161的氨基酸序列高度保守,而位于该保守区的活性残基Cys154、Cys158对GAPDH酶活性及调节GAPDH参与非糖酵解代谢过程至关重要[10]。在拟南芥中,细胞质AtGAPC1活性严格依赖Cys149,该位点的突变导致该酶几乎无活性,而残基Cys153的突变对酶活性影响甚微[16]。小麦细胞质GAPDH底物(G3P和NAD+)结合试验表明,Cys154可能至少是受氧化作用的活性位点之一,该研究与在人类或拟南芥GAPDH同源酶中的发现相同[19]。GAPDH蛋白活性位点的Cys残基巯基容易受生理或体外H2O2、NO、GSH等作用发生氧化还原修饰,如磺酸化、硝基化和谷胱甘肽化等,导致GAPDH蛋白可逆或不可逆失活,而失活的GAPDH通过与其他蛋白相互作用改变其亚细胞定位,参与不同生理过程[12-13,17-18]。
本研究在获得小麦细胞质TaGAPC基因cDNA全长序列的基础上,采用重叠延伸PCR法对其催化活性位点的Cys154、Cys158残基分别进行定点突变,连接pGEM-T easy克隆载体进行测序等验证突变成功,并且未引入其他突变位点。然后构建了原核表达载体pET28a(+)-TaCys154S/TaCys158S,并采用镍柱纯化技术获得可溶性重组蛋白,为后续TaGAPC的Cys154、Cys158这2个重要半胱氨酸活性位点分析及研究TaGAPC在小麦细胞中参与的调控机制提供试验材料。本研究在TaGAPC基因中引入突变位点时采用了重叠延伸PCR技术。该技术运用广泛、灵活,不受突变位置及突变类型的限制,可对DNA片段进行定点突变甚至多点突变,在阐明基因的调控机理、改造蛋白质结构等分子生物学领域中具有潜在的应用价值[22]。
参考文献:
[1]Danshina P V,Schmalhausen E V,Avetisyan A V,et al.Mildly oxidized glyceraldehyde-3-phosphate dehydrogenase as a possible regulator of glycolysis[J].IUBMB Life,2001,51(5):309-314.
[2]Zhang X H,Rao X L,Shi H T,et al.Overexpression of a cytosolic glyceraldehyde-3-phosphate dehydrogenase geneOsGAPC3 confers salt tolerance in rice[J].Plant Cell,Tissue and Organ Culture (PCTOC),2011,107(1):1-11.
[3]Kubo H.Cloning and expression analysis of putative glyceraldehyde-3-phosphate dehydrogenase genes inPiloboluscrystallinus[J].Mycoscience,2011,52(2):99-106.
[4]Wu Y,Wu M,He G,et al.Glyceraldehyde-3-phosphate dehydrogenase:a universal internal control for western blots in prokaryotic and eukaryotic cells[J].Analytical Biochemistry,2012,423(1):15-22.
[5]Sirover M A.Role of the glycolytic protein,glyceraldehyde-3-phosphate dehydrogenase,in normal cell function and in cell pathology[J].Journal of Cellular Biochemistry,1997,66(2):133-140.
[6]Sirover M A.New nuclear functions of the glycolytic protein,glyceraldehyde-3-phosphate dehydrogenase,in mammalian cells[J].Journal of Cellular Biochemistry,2005,95(1):45-52.
[7]Sirover M A.Subcellular dynamics of multifunctional protein regulation:mechanisms of GAPDH intracellular translocation[J].Journal of Cellular Biochemistry,2012,113(7):2193-2200.
[8]Sirover M A.On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase:biochemical mechanisms and regulatory control[J].Biochimica et Biophysica Acta,2011,1810(8):741-751.
[9]付国良,黄晓红.甘油醛-3-磷酸脱氢酶功能的研究进展[J].生物物理学报,2013,29(3):181-191.
[10]Zaffagnini M,Fermani S,Costa A,et al.Plant cytoplasmic GAPDH:redox post-translational modifications and moonlighting properties[J].Frontiers in Plant Science,2013,4:450.
[11]Zaffagnini M,Bedhomme M,Marchand C H,et al.Redox regulation in photosynthetic organisms:focus on glutathionylation[J].Antioxidants & Redox Signaling,2012,16(6):567-586.
[12]Cejudo F J,Meyer A J,Reichheld J P,et al.Thiol-based redox homeostasis and signaling[J].Frontiers in Plant science,2014,5:266.
[13]卢倩,弭晓菊,崔继哲.植物甘油醛-3-磷酸脱氢酶作用机制的研究进展[J].生物技术通报,2013(8):1-6.
[14]Holtgrefe S,Gohlke J,Starmann J,et al.Regulation of plant cytosolic glyceraldehyde 3-phosphate dehydrogenase isoforms by thiol modifications[J].Physiologia Plantarum,2008,133(2):211-228.
[15]Zaffagnini M,Morisse S,Bedhomme M,et al.Mechanisms of nitrosylation and denitrosylation of cytoplasmic glyceraldehyde-3-phosphate dehydrogenase fromArabidopsisthaliana[J].The Journal of Biological Chemistry,2013,288(31):22777-22789.
[16]Bedhomme M,Adamo M,Marchand C H,et al.Glutathionylation of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the model plantArabidopsisthalianais reversed by both glutaredoxins and thioredoxinsinvitro[J].The Biochemical Journal,2012,445(3):337-347.
[17]Guo L,Devaiah S P,Narasimhan R,et al.Cytosolic glyceraldehyde-3-phosphate dehydrogenases interact with phospholipase Dδ to transduce Hydrogen peroxide signals in theArabidopsisresponse to stress[J].The Plant Cell,2012,24(5):2200-2212.
[18]Morigasaki S,Shimada K,Ikner A,et al.Glycolytic enzyme GAPDH promotes peroxide stress signaling through multistep phosphorelay to a MAPK cascade[J].Molecular Cell,2008,30(1):108-113.
[19]Piattoni C V,Guerrero S A,Iglesias A A.A differential redox regulation of the pathways metabolizing glyceraldehyde-3-phosphate tunes the production of reducing power in the cytosol of plant cells[J].International Journal of Molecular Sciences,2013,14(4):8073-8092.
[20]Ho S N,Hunt H D,Horton R M,et al.Site-directed mutagenesis by overlap extension using the polymerase chain reaction[J].Gene,1989,77(1):51-59.
[21]罗师平,冷希岗.基于PCR的体外诱变技术[J].国外医学:生物医学工程分册,2005,28(3):188-192.
[22]何震宇,李月琴,林元藻.重叠延伸PCR对DNA片段进行定点双突变[J].氨基酸和生物资源,2007,29(3):78-82.
[23]Piattoni C V,Rius S P,Gomez-Casati D F,et al.Heterologous expression of non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase fromTriticumaestivumandArabidopsisthaliana[J].Biochimie,2010,92(7):909-913.
[24]张霞,王艳,张富春.逆境胁迫下甘油醛-3-磷酸脱氢酶功能多元化的研究进展[J].植物生理学报,2013,49(1):24-28.
Construction ofTaGAPCSite-directed Mutagenesis Gene Vector and Prokaryotic Expression Analysis from Wheat
GUO Ziping1,ZHAI Qinghua1,ZENG Lingfeng1,DENG Xiping2,YANG Shushen1
(1.College of Life Sciences,Northwest A&F University,Yangling712100,China;2.State Key Laboratory of Soil Erosion and Dryland Farming on Loess Plateau,Northwest A&F University,Yangling712100,China)
Abstract:In order to elucidate the roles of catalytic active Cys154,Cys158in Triticum aestivum L.cytoplasmic glyceraldehyde-3-phosphate dehydrogenase (TaGAPC),these two cysteins were site-directed mutated into serine by overlap-extension PCR.Then recombinant prokaryotic expression vectors of TaGAPC and its mutants TaCys154S/TaCys158S were constructed and transformed into E.coli BL21.The recombinant protein were induced by 0.25 mmol/L IPTG at 25 ℃ and subsequently purified by Ni2+-IDA column.These researches could lay the foundation for the enzyme activity assay of TaGAPC and its mutants TaCys154S/TaCys158S and cysteine active sites.
Key words:TaGAPC;Overlap-extension PCR;Site-directed mutagenesis;Prokaryotic expression;Protein purification
doi:10.7668/hbnxb.2016.01.008
中图分类号:Q78;S512.03
文献标识码:A
文章编号:1000-7091(2016)01-0046-05
作者简介:郭子平(1989-),男,河南商丘人,在读硕士,主要从事植物逆境细胞分子生物学研究。通讯作者:杨淑慎(1956-),女,陕西黄陵人,教授,博士,主要从事植物抗逆生理、植物细胞培养工程方面的研究。
基金项目:国家自然科学基金项目(31271625);黄土高原土壤侵蚀与旱地农业国家重点实验室专项 (10502)
收稿日期:2015-11-27
猜你喜欢
中国中药杂志(2017年2期)2017-03-25 17:19:57
江苏农业科学(2016年10期)2017-02-05 01:33:24
江苏农业科学(2016年7期)2016-10-20 01:13:21
江苏农业科学(2016年7期)2016-10-20 01:11:25
湖南大学学报·自然科学版(2016年6期)2016-07-14 09:18:03
江苏农业科学(2016年4期)2016-06-14 01:46:43
江苏农业科学(2015年9期)2015-10-20 21:04:29
江苏农业科学(2015年7期)2015-08-20 20:58:46
江苏农业科学(2015年3期)2015-07-31 17:10:52
热带农业科学(2015年3期)2015-04-28 04:29:09
