
建立液相色谱-质谱串联法测定大鼠微透析样品中美罗培南的浓度
2016-01-31 06:51周静超郭明星童卫杭
解放军医药杂志 2015年12期
彭 珑,王 丹,吴 诚,周静超,郭明星,童卫杭
建立液相色谱-质谱串联法测定大鼠微透析样品中美罗培南的浓度
彭珑,王丹,吴诚,周静超,郭明星,童卫杭

[摘要]目的建立高效液相色谱-质谱串联法(HPLC-MS/MS)用于监测微美罗培南在透析液中实时变化的浓度,为应用微透析技术对感染靶部位美罗培南的药动/药效学研究奠定基础。方法采用COSMOSIL 5C18-PAQ(150 mm×4.6 mm, 5 μm)色谱柱进行分离,流动相为乙腈-0.1%甲酸水,梯度洗脱,流速1.0 ml/min,分析时间为8 min;美罗培南和内标(安替比林)均在ESI正离子模式扫描,以多反应监测(MRM)模式进行扫描,检测离子对美罗培南m/z 384.2→141.2,内标m/z 189.2→104.0。结果美罗培南在0.01~40 μg/ml线性范围内线性关系良好(r=0.9979);日内及日间精密度在9.76%以内,准确度-7.87~0.28%。结论本法灵敏度高、专属性强、精密度好、准确度高,适用于微透析样品的快速有效分析。
[关键词]高效液相色谱-串联质谱技术;美罗培南;微透析;大鼠
脓毒症(sepsis)是由感染引起的全身炎症反应综合征(systemic inflammatory response syndrome, SIRS),是严重创伤、休克、感染、外科大手术常见并发症,进一步发展可导致脓毒性休克、多器官功能障碍综合征(multiple organ dysfunction syndrome,MODS)。脓毒症来势凶猛、病情进展迅速、预后险恶,据国外流行病学调查显示脓毒症病死率高达30%~70%[1-2],已超过心肌梗死,是重症监护病房内非心脏病患者死亡的主要原因。“早期、强效、广谱”给予抗生素是有效控制脓毒症的关键。碳青霉烯类抗菌药物是抗菌谱广、抗菌活性强的抗生素,其对大多β-内酰胺酶稳定、毒性低,对广谱青霉素和第2、3、4代头孢菌素耐药性细菌有活性,且细菌对该类药物不存在交叉耐药等特点,成为治疗脓毒症感染的主要抗菌药物,美罗培南是临床常用药物之一。多中心研究表明美罗培南对于并发感染的住院患者具有良好的临床疗效且耐受性好[3-5]。
然而,脓毒症疾病发生发展中的一系列病理生理变化对抗生素给药方案产生了复杂影响[6],目前无法准确监测感染靶部位药物浓度是导致抗生素治疗脓毒症失败的主要原因,微透析技术是一项新型的生物活体取样技术,可以直接对感染靶部位进行取样。本研究旨在建立一种定量方法用于测定微透析液中美罗培南的浓度,为应用微透析技术对感染靶部位美罗培南的药动/药效学研究奠定基础。前期已有部分文献报道过美罗培南在血浆中的定量方法主要包括高效液相色谱-紫外检测器联用(HPLC-UV)[6-11]和高效液相色谱-质谱联用技术(HPLC-MS/MS)[12-14],对于美罗培南在其他体液中如脑脊液中的药物浓度也有报道,一般也是应用HPLC-UV法[15]。由于HPLC-UV法灵敏度较低,不适于微透析样本的测定,而HPLC-MS/MS法不仅灵敏度高且专属性强,适用于样本量少,含量较低的微透析样品[16]。因此,本实验拟建立大鼠微透析样本中美罗培南的HPLC-MS/MS测定方法,并进行全面的方法学验证与评估。
1材料与方法
1.1实验动物选用SPF级雄性SD大鼠6只,购自北京维通利华公司,体重为300~350 g,月龄4~5个月。饲养于本机构动物室:温度(23±2)℃,湿度(60±5)%,12 h昼夜循环光照,饲养期间自由饮水和摄食。大鼠适应环境1周后开始实验。动物合格证编号:SCXK(京)2012-0001。
1.2仪器与试剂安捷伦1100高效液相色谱仪(美国Agilent公司产品),配有四元输液泵、自动进样器及切换阀;AB 3200 QTrap型质谱仪(美国Applied Biosystems公司产品),配有电喷雾离子源(ESI)以及Analyst 1.4.1数据处理软件;CMA 150型动物保温垫、CMA 402型微量注射泵、MAB 85型低温样品收集器(均购自美国CMA公司)。美罗培南对照品(批号130506-200702,含量87.0%)、安替比林对照品(批号100506-200301),均购自中国药品生物制品检定所;乙腈、甲醇均为色谱纯(购自美国Fisher公司);甲酸为分析纯(购自美国Fisher公司);超纯水通过本实验室Millipore Synergy UV型超纯水机获得;水合氯醛购自中国人民解放军第306医院(批号140107);注射用美罗培南及复方氯化钠注射液,购自华润双鹤药业股份有限公司,批号A201409041。
1.3试验条件
1.3.1色谱条件:色谱柱:COSMOSIL 5C18-PAQ柱(150 mm×4.6 mm, 5 μm);流动相:0.1%甲酸水(A)-乙腈(B),梯度洗脱程序见表1;流速为1.0 ml/min,分流比1∶1;柱温20℃,进样室温度4℃;进样量:5 μl;分流比1∶1。

表1 流动相梯度洗脱程序
1.3.2质谱条件:采用电喷雾电离源(ESI),正离子模式扫描;离子喷射电压:5500 V;离子源温度:500℃;源内气体1(GS1,N2)压力:20 kPa;气体2(GS2,N2)压力:30 kPa;气帘气体(N2)压力:10 kPa;扫描方式:多反应监测(MRM);碰撞气(N2)压力:Medium;用于定量分析的离子对分别为美罗培南m/z 384.2→141.2,解簇电压(DP) 24 V,碰撞能量(CE) 20 eV;内标安替比邻m/z 189.2→104.0,DP为57 V,CE为22 eV。美罗培南、内标的二级质谱图见图1。
1.4储备液、标准溶液及质控样品的制备精密称取美罗培南对照品10 mg置于10 ml容量瓶中,用甲醇溶解并稀释至刻度,配制成浓度为1.0 mg/ml的美罗培南储备液,存于4℃,备用。将储备液用复方氯化钠注射液依次稀释至系列浓度的标准曲线溶液,浓度依次为40、10、5、1、0.5、0.1、0.05、0.01 μg/ml。同法制备质控(QC)样品,高中低浓度分别为30.0、0.50、0.025 μg/ml。同样精密称取内标安替比邻对照品适量置于容量瓶中,用甲醇溶解并稀释至刻度,得到其储备液。再用复方氯化钠注射液将储备液稀释至0.8 μg/ml,即得内标溶液,存于4℃,备用。
1.5微透析样品收集与前处理肺组织微透析样品收集:水合氯醛将SD大鼠麻醉(0.35 ml/100 g),固定在37℃保温垫上,腹部朝上。眼科剪将大鼠腹部剪开,使肺部暴露,将线性探针平行植入肺部组织,再将探针与微透析装置相连。以10 μl/ml的流速泵入复方氯化钠注射液,排除气泡并冲洗管路,再以1 μl/ml的流速继续泵入复方氯化钠注射液30 min以达到平衡,并收集空白透析液;将用生理盐水溶解的注射用美罗培南给予大鼠尾静脉注射后,以冷冻样品收集器连续收集透析液,温度为4℃,采样间隔为20 min,连续采集6 h。微透析样品前处理:精密取肺组织微透析样品20 μl,加入内标20 μl,涡旋混匀,以14 000 r/min,离心5 min,取上清液5 μl用于LC-MS/MS分析。
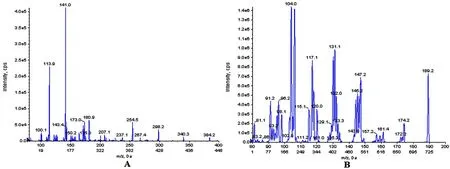
图1 美罗培南及内标的二级质谱图(A:美罗培南;B:安替比邻)
1.6方法学验证
1.6.1方法的专属性:分别取6只大鼠的肺组织空白透析液,将空白透析液的色谱图与空白透析液中加入美罗培南对照品和内标处理后得到的色谱图,以及给药后大鼠透析液的色谱图进行比较,看大鼠空白透析液中其他内源性成分对待测成分是否有干扰,以及给药后是否会产生代谢产物对美罗培南的测定有干扰。
1.6.2标准曲线及最低定量限(LLOQ):取系列浓度的标准曲线溶液各20 μl,分别加入内标液20 μl,涡旋混匀,以14000 r/min,离心5 min,取上清液5 μl用于LC-MS/MS分析,建立美罗培南的标准曲线。以美罗培南的浓度x为横坐标,以其与内标的峰面积比y为纵坐标,用加权最小二乘法(W=1/X2)计算回归方程及相关系数(r),要求r值大于0.99。LLOQ即为标准曲线的最低浓度点,平行处理6份,精密度需小于20%,准确度在±20%范围内。
1.6.3精密度和准确度:取高、中、低3个浓度的QC样品,每个浓度平行制备6份,进样分析,计算日内精密度和准确度,重复操作,连续测定3 d并随行当日标准曲线,计算日间精密度和准确度。以相对误差(RE)表示准确度,相对标准偏差(RSD)表示精密度。要求高、中浓度QC样品RSD≤15%,RE在±15%范围内,低浓度QC样品RSD≤20%,RE在±20%范围内。
1.6.4稳定性:取高、中、低浓度QC样品于制备好后在20℃下放置4、8、12 h后进样分析,考察样品处理后在室温下放置12 h的稳定性;将处理后的样品在4℃下放置4、8、12及24 h后进样分析,考察样品处理后在进样条件下放置24 h的稳定性;将样品在-40℃下冷冻,经24 h后取出至室温自然融解,再置于-40℃下冷冻,考察经3个冻融循环后的稳定性;将质控样品在37℃下放置3、6、9、12 h后处理进样分析,考察样品在37℃的透析过程中的稳定性。
2结果
2.1分析方法确证专属性对比图2A、2B,可以看出,在美罗培南和内标的出峰处没有干扰,说明大鼠空白透析液中没有内源性成分的干扰;对比图2B和图2C可以看出,在给药后的大鼠微透析液中也没有代谢物的干扰,说明本方法专属性良好。
2.2分析方法确证标准曲线及最低定量限(LLOQ)通过标准曲线样品的浓度和峰面积求得线性回归方程:Y=0.000491X+0.00163,相关系数r=0.9979,说明美罗培南在0.01~40 μg/ml线性范围内线性关系良好,最低定量限为标曲的最低点,即0.01 μg/ml,将此浓度点的样品平行处理6份,进样分析,精密度为7.25%,准确度为-4.81%,在合格范围内。
2.3分析方法确证精密度和准确度分析方法的日内及日间精密度和准确度实验结果见表2。美罗培南日内和日间精密度分别在5.86%~9.76%和2.41%~5.23%范围内,准确度-7.87%~0.28%,结果表明分析方法的准确度和精密度均较好。
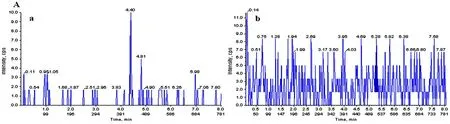
图2 LC-MS/MS法测定大鼠微透析液中美罗培南(a)及内标(b)的色谱图A为空白透析液色谱图;B为空白透析液中加入美罗培南0.01 μg/ml(LLOQ)以及内标0.8 μg/ml处理后的色谱图;C为大鼠尾静脉注射美罗培南注射液1 h时肺组织透析液的色谱图
表2 美罗培南测定方法的精密度和准确度±s)
2.4分析方法确证稳定性样品稳定性结果见表3。从结果可见,透析样品处理后在20℃下放置12 h、在4℃下放置24 h以及在37 ℃下的透析环境中放置12 h后,样品均保持稳定,未发生明显的降解反应;将样品在-40℃下经过3个冻融循环后的仍保持稳定。
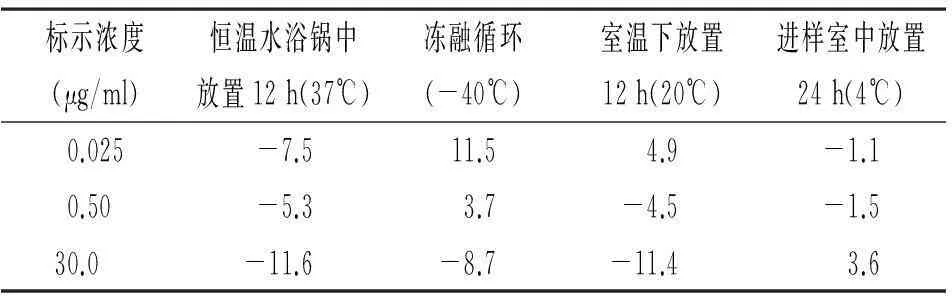
表3 美罗培南微透析样品的稳定性(RE%)
3讨论
质谱条件的选择,建立LC-MS/MS检测方法的第一步即是选择美罗培南和内标的母子离子对,用于MRM模式监测。用甲醇分别将美罗培南和内标储备液稀释至1 μg/ml,以5 μl/min的流速直接进质谱分析。采用ESI正离子模式,美罗培南及内标的母离子均为[M+H]+峰,质荷比分别为m/z 384.2和189.2。根据子离子选择原则,每个化合物的母离子应选择2个子离子来确证目标化合物,但选择响应更强的作为定量离子对,因此最终选择美罗培南和内标用于定量的离子对分别为m/z 384.2→141.2和m/z 189.2→104.0。
色谱条件的选择,为了获得良好的峰型、较高的灵敏度、较短的分析时间,我们对流动相的选择、比例、柱温等均进行了优化。有报道称在流动相里加入少量的电解质即能显著改善电离效率,从而提高化合物的响应灵敏度[17]。因此,我们比较了在水相中加入醋酸铵和甲酸作为流动相的色谱图,发现以甲酸水作为水相,美罗培南和内标的峰形均较好,且离子强度比以醋酸铵水作为水相时更强,另外还优化了加入甲酸的量,认为0.1%的甲酸水已能使两化合物具有良好的峰形和响应;分别以甲醇和乙腈作为有机相也进行了比较,结果表明,以乙腈作为有机相的灵敏度比甲醇稍高,且峰型更好,保留时间提前;因此,最终选定乙腈-0.1%甲酸水作为本次实验的流动相。另外,为了获得更窄的色谱峰型和更快的分析时间,采用了梯度洗脱。比较了20、30、40℃柱温条件下的色谱峰,发现并没有太大差异,且柱温从20℃升至40℃,保留时间仅提前了0.1 min,因此,最终选择20℃柱温进行实验。
实验中的样品均为微透析样品,复方氯化钠注射液中含各种无机盐离子,而由于无机盐不具有挥发性而不能进质谱,因此,在液相方法设定中,从0~2.5 min即一个柱体积切换至废液,从2.51 min才开始进质谱,由于盐离子在色谱柱上不会有保留,因此一个柱体积已能将盐离子全部除去,从而保护质谱离子源和锥孔不受污染,并可提高灵敏度,目前很多微透析-液质联用技术都是采用这种方法来减少或避免非挥发性盐离子或小分子物质对质谱的影响[18~19]。
在本实验中,建立并验证了LC-MS/MS法以测定大鼠尾静脉注射美罗培南注射液后肺部组织透析液中美罗培南的浓度。此法灵敏度高、专属性强、精密度好、准确度高、分析时间短,便于透析样品的快速有效分析,并可进一步应用于血管、肌肉、皮肤其他组织的的微透析实验中。
[参考文献]
[1]Shapiro N, Howell M D, Bates D W,etal. The association of sepsis syndrome and organ dysfunction with mortality in emergency department patients with suspected infection[J].Ann Emerg Med, 2006,48(5):583-590.
[2]Kleinpell R M, Graves B T, Ackerman M H. Incidence pathogenesis and management of sepsis an overview[J].AACN Adv Crit Care, 2006,17(4): 385-393.
[3]Cox C E, Holloway W J, Geckler R W. A multicenter comparative study of meropenem and imipenem/cilastatin in the treatment of complicated urinary tract infections in hospitalized patients[J].Clin Infect Dis, 1995,21(1):86~92.
[4]Kollef M H. Update on the appropriate use of meropenem for the treatment of serious bacterial infections[J].Clin Infect Dis, 2008,47(Suppl1):S1-2.
[5]Zhanel G G, Wiebe R, Dilay L,etal. Comparative review of the carbapenems[J].Drugs, 2007,67(7):1027-1052.
[6]Roberts J A, Lipman J. Pharmacokinetic issues for antibiotics in the critically ill patient[J].Crit Care Med, 2009,37(3):840-851.
[7]Denooz R, Charlier C. Simultaneous determination of five beta-lactam antibiotics (cefepim, ceftazidim, cefuroxim, meropenem and piperacillin) in human plasma by high-performance liquid chromatography with ultraviolet detection[J].J Chromatogr B Analyt Technol Biomed Life Sci, 2008,864(1-2):161-167.
[8]Dailly E, Bouquié R, Deslandes G,etal. A liquid chromatography assay for a quantification of doripenem, ertapenem, imipenem, meropenem concentrations in human plasma: application to a clinical pharmacokinetic study[J].J Chromatogr B Analyt Technol Biomed Life Sci, 2011,879(15-16):1137-1142.
[9]Briscoe S E, McWhinney B C, Lipman J,etal. A method for determining the free (unbound) concentration of ten beta-lactam antibiotics in human plasma using high performance liquid chromatography with ultraviolet detection[J].J Chromatogr B Analyt Technol Biomed Life Sci, 2012,907:178-184.
[10]Jamal J A, Mat-nor M B, Mohamad-nor F S,etal. Pharmacokinetics of meropenem in critically ill patients receiving continuous venovenous haemofiltration: A randomised controlled trial of continuous infusion versus intermittent bolus administration[J].Int J Antimicrob Agents, 2015,45(1):41-45.
[11]Casals G, Hernández C, Hidalgo S,etal. Development and validation of a UHPLC diode array detector method for meropenem quantification in human plasma[J].Clin Biochem, 2014,47(16-17):223-227.
[12]Ohmori T, Suzuki A, Niwa T,etal. Simultaneous determination of eight β-lactam antibiotics in human serum by liquid chromatography-tandem mass spectrometry[J].J Chromatogr B Analyt Technol Biomed Life Sci, 2011,879(15-16):1038-1042.
[13]Carlier M, Stove V, Roberts J A,etal. Quantification of seven β-lactam antibiotics and two β-lactamase inhibitors in human plasma using a validated UPLC-MS/MS method[J].Int J Antimicrob Agents, 2012,40(5):416-422.
[14]Colin P, De Bock L, T'jollyn H,etal. Development and validation of a fast and uniform approach to quantify β-lactam antibiotics in human plasma by solid phase extraction-liquid chromatography-electrospray-tandem mass spectrometry[J].Talanta, 2013,103:285-293.
[15]Morita A, Kamei S, Minami M,etal. Open-label study to evaluate the pharmacodynamics clinical efficacy and safety of meropenem for adult bacterial meningitis in Japan[J].J Infect Chemother, 2014,20(9):535-540.
[16]De Andrade C, De Araújo Lock G, Pigatto M C,etal. Validation of LC-MS/MS method applied to evaluation of free tissue concentrations of vildagliptin in diabetic rats by microdialysis[J].Biomed. Chromatogr, 2014,28(12):1722-1727.
[17]Zhao Y, Sun Y, Li C. Simultaneous determination of Ginkgo flavonoids and terpenoids in plasma: Ammonium formate in LC mobile phase enhancing electrospray ionization efficiency and capacity[J].Journal of the American Society for Mass Spectrometry, 2008,19(3):445-449.
[18]LI T F, Xue W, Li M,etal. Establishment of an LC-MS/MS method to determine content of six amino acids in rat brain dialysate[J].Chin New Drug J, 2015,24(14):1659-1664.
[19]Greco S, Danyszb W, Zivkovicc A,etal. Microdialysate analysis of monoamine neurotransmitters-a versatile and sensitive LC-MS/MS method[J].Analytica Chimica Acta, 2013,771(7):65-72.
(收稿时间:2015-09-22修回时间:2015-10-25)
·论著·

LC-MS/MS Method in Determination of Meropenem Concentration of Microdialysis Samples of Rats
PENG Long, WANG Dan, WU Cheng, ZHOU Jing-chao, GUO Ming-xing, TONG Wei-hang ( Department of Pharmacy, General Hospital of the Second Artillery of Chinese PLA, Beijing 100088, China)
[Abstract]ObjectiveTo establish a high performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) method to monitor real-time concentration changes of Meropenem during microdialysis samples of rats so as to provide the foundation for further pharmacokinetic/pharmacodynamic (PK/PD) study of Meropenem on infected tissues with microdialysis technique. MethodsChromatographic separation was performed on a COSMOSIL 5C18-PAQ (150 mm×4.6 mm, 5 μm) column by a gradient elution, the mobile phase was composed of acetonitrile and -0.1% formic acid aqueous solution at a 1.0 mL/min flow rate in a run time of 8 min. The quantification of Meropenem and internal standard (Antipyrine) were determined by multiple reaction monitoring (MRM) mode with a positive electrospray ionization (ESI), and the ion transitions were m/z 384.2→141.2 and m/z 189.2→104.0 for Meropenem and internal standard respectively. ResultsThe Meropenem showed good linear correlation in a range of 0.01-40 μg/mL (r=0.9979). The intra- and inter-day precisions [relative standard deviation (RSD) %] were all within 9.76%, and accuracy (RE%) ranged from -7.87% to 0.28%. ConclusionThe established LC-MS/MS method of Meropenem is accurate with good sensitivity, specificity and precision, which can be applied in quick and effective analysis of Meropenem concentration in microdialysis samples.
[Key words]High performance liquid chromatography-tandem mass spectrometry; Meropenem; Microdialysis; Rats
[DOI]10.3969/j.issn.2095-140X.2015.12.024
[文献标志码][中国图书资料分类号]R446.1 R943A
[文章编号]2095-140X(2015)12-0103-06
[通讯作者]童卫杭,E-mail:sonatawh@163.com
[基金项目][作者单位]100088 北京,解放军第二炮兵总医院药学部
猜你喜欢
中国典型病例大全(2022年7期)2022-04-22
中国医院用药评价与分析(2018年6期)2018-07-12
中国感染与化疗杂志(2017年4期)2017-01-15
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
科学与财富(2016年28期)2016-10-14
中国卫生标准管理(2015年3期)2016-01-15
中国药业(2014年12期)2014-06-06
中国药业(2014年21期)2014-05-26
