
鸭坦布苏病毒E基因的分子特性分析
2015-11-03 03:26邓明俊郑小龙
中国动物检疫 2015年12期
孙 涛,王 超,邓明俊,王 群,郑小龙,徐 彪
(1.山东出入境检验检疫局,山东青岛 266002;2.泰安出入境检验检疫局,山东泰安 250014)
鸭坦布苏病毒E基因的分子特性分析
孙 涛1,王 超2,邓明俊1,王 群1,郑小龙1,徐 彪1
(1.山东出入境检验检疫局,山东青岛 266002;2.泰安出入境检验检疫局,山东泰安 250014)
为给鸭坦布苏病毒(duck tembusu virus,DTMUV)囊膜糖蛋白选择良好的抗原域和宿主表达系统提供参考依据,本研究通过选取DTMUV QD株基因文库中的一个重组质粒并测序,结合NCBI的ORF Finder和BLAST工具发现一个全长为1488 bp完整开放阅读框架(Open Reading Frame,ORF)片段。将该ORF编码蛋白通过NCBI的BLASTP分析和DNAstar进化树分析,确定该蛋白与类黄病毒E基因编码的蛋白具有较高的相似性。随后应用生物信息学分析工具Conserved Domains确定保守结构域、运用SignalP4.1预测信号肽、TMHMM 2.0预测跨膜区。应用在线程序NetNGlyc1.0预测糖基化位点,NetPhos2.0预测磷酸化位点、ProtScale进行疏水性分析。最后运用在线EMBOSS和自动蛋白同源建模数据库进行3D结构预测以及密码子偏爱性分析。结果表明,E蛋白与其它黄病毒衣壳蛋白具有相似的功能,没有信号肽切割位点,在451-468和475-492aa区域含有跨膜区,为相对低表达基因,含有较多种类的稀有密码子,与人密码子使用频率较为接近。这为进一步研究DTMUV E基因的体外表达和宿主选择提供了分子生物学依据。
鸭坦布苏病毒;生物学信息学;结构预测
鸭源坦布苏病毒病毒(duck tembusu virus,DTMUV)属黄病毒科黄病毒属成员[1,2],是引起国内以卵巢炎为特征[3-5]的水禽新发传染性疾病的主要病原。该病流行广泛、传播迅速,自2010年首次报道以来[6],该病毒已感染产蛋鸭、肉鸭和鹅等养殖水禽,并造成其产蛋严重下降。
为建立该病毒的快速检测方法,对其主要结构蛋白基因进行表达,鉴于前期表达蛋白的密码子偏爱性和抗原结构域尚处未知状态,本实验室拟根据核酸文库中的重组质粒测序结果,初步确定DTMUV毒株的囊膜糖蛋白E基因的序列。再对该基因编码蛋白进行生物信息学分析,查找主要抗原结构域,预测蛋白结构,并通过EMBOSS(The European Molecular Biology Open Software Suite)软件对上述基因序列进行密码子偏爱性分析,将结果与大肠杆菌、酵母、及人的密码子偏爱性(数据来自Codon usage database)进行比较,为选择合适的表达系统提供依据。
1 材料与方法
1.1材料
1.1.1基因文库。DTMUV QD株基因文库,由青岛蔚蓝生物有限公司构建。
1.1.2主要生物信息学分析软件。DNAStar6.0软件、NCBI ORF Finder工具、BLASTN工具、BLASTP工具、Conserved Domains查找工具,在线TMHMM程序、Signal P4.1程序、NetNGlyc1.0程序、NetPhos2.0程序、ProtScale程序、在线3D建模程序、在线EMBOSS的CHIPS程序、CUSP程序。
1.2分析方法
1.2.1核酸序列的分子特性分析。选取构建的DTMUV QD株 DNA基因文库中的一个重组质粒,送上海生物工程公司测序,并对测定的重组质粒DNA序列运用NCBI ORF Finder工具,进行网上在线开放阅读框架(Open Reading Frames,ORF)的寻找和分析,确定DTMUV E基因的ORF。将所测序列利用NCBI的BLASTN工具进行核酸序列的相似性搜索,然后应用DNAstar6.0软件MegAlign程序中的Clustal V工具进行核酸序列同源性分析。
1.2.2蛋白质序列的分子特性分析。运用瑞士蛋白质专家网在线翻译工具将目标核酸序列翻译成氨基酸序列,然后对该蛋白组分进行分析:(1)运用瑞士蛋白质分析专家系统(http:// expasy.org/cgi-bin/protparam)将目标核酸序列翻译成氨基酸序列,然后分析该蛋白的基本理化性质;并运用Conserved Domains查找工具分析该基因的结构域;(2)运用在线系统(http:// www.cbs.dtu.dk/services/) 中 的 TMHMM 2.0 、SignalP4.1、NetNGlyc1.0、和NetPhos2.0程序[7],分析DTMUV E蛋白潜在的跨膜区域、信号肽、N-糖基化位点和磷酸化位点;(3)运用瑞士蛋白质分析专家系统(http://www.expasy.org/cgi-bin/ protscale.pl)中的ProtScale 程序,工具分析蛋白的疏水性 ;(4)运用在线软件(http://www.rcsb.org/ pdb/explore/)的3D建模程序,预测蛋白二级结构和三级结构[8]。
1.2.3DTMUV E基因的密码子偏爱性分析方法。采用EMBOSS在线分析系统(http://emboss. toulouse.inra.fr/)中的CHIPS、CUSP程序[9-10],对E基因进行密码子使用偏爱性分析。采用CHIPS程序模块计算序列中有效密码子的ENC(Effective Number of Codons)值[11]。该值是一个基因的密码子使用频率与同义密码子平均使用频率偏差的量化值。范围在20(每个氨基酸只使用一个密码子的极端情况)到61(各个密码子均被平均使用)时可单独由密码子使用数据计算得出,与基因长度及氨基酸组成无关,可对基因的密码子偏爱性程度提供一个客观的评判标准。用CUSPS程序计算密码子频率表,分析得到DTMUV基因密码子使用频率(1/1000代表该密码子在编码基因中出现的频率),并与大肠杆菌、酵母和人的密码子使用频率(数据来自Condon usuage database,http:www.kazusa. or.jp/condon)比较,看密码子偏爱性模式与三种表达系统的相似度。
2 结果
2.1核酸序列的分子特性分析
经对DTMUV QD株 cDNA基因文库中一个重组质粒的测序和NCBI ORF Finder及BLASTN工具分析得到编码该病毒E基因的一个由1 488个碱基组成的完整ORF(图1),该ORF编码的蛋白与衣壳蛋白家族相关并与类黄病毒表面糖蛋白基因有很高的相似性,编码蛋白属于衣壳糖蛋白家族成员。
通过核苷酸序列相似性搜索,结果(图2)发现本株病毒的阅读框与目标核苷酸序列(JF895923.2)完全相似的序列高达99%。同时,通过进一步比对,发现它们的序列之间只有一个碱基的差异。另外,Clustal V method工具对DTMUV E基因的核酸序列以及Genbank上6个同属黄病毒科的不同黄病毒属的同源基因进行核酸序列的同源性比对,系统进化树分析结果(图3)可见:本株病毒编码的E基因与黄病毒科成员类聚,与黄热病毒(Yellow fever virus)、登革热病毒(Dengue virus)、伊利乌斯脑炎病毒(Ilheus virus)、日本脑炎病毒(Japanese encephalitis virus)、西尼罗病毒(West Nile virus)、巴格扎病毒(Bagaza virus)和坦布苏病毒(Tembusu virus)具有遗传进化关系。其中与黄热病毒(Yellow fever virus)亲缘关系最远,与巴格扎病毒(Bagaza virus)亲缘关系较近但遗传距离介于病毒种的水平上。由此可见,DTMUV E蛋白与其它类黄病毒既有高度的同源性,又存在一定的差异。
2.2蛋白质序列的分子特性分析
2.2.1E蛋白基本理化性质分析。瑞士蛋白质专家网在线翻译工具预测DTMUV E基因的ORF编码一条由494个氨基酸残基组成的多肽,相对分子量为53.6 KDa,等电点理论值为7.14,分子式为C2378H3717N637O713S31。其氨基酸组成成分如表1所示。进一步分析表明,蛋白质中含量最高的氨基酸为Gly(10.3%)、Val(9.1%)和Ala(7.3%);而含量最少的氨基酸为Trp(2.0%)、Cys(2.2%)、Tyr(2.8%)和Arg(3.2%)。
2.2.2E 蛋白结构域分析。NCBI Conserved Domains查找工具分析发现该基因氨基酸序列与黄病毒科E基因类似,含有3个保守结构域 (图4),说明E蛋白与其它黄病毒衣壳蛋白可能具有相似的功能。

图1 ORF Finder的“SixFrames”分析结果

图2 DTMUV E基因与鸭坦布苏病毒组JF895923.2的序列比对结果

图3 DTMUV与参考毒株同源基因氨基酸序列进化树分析
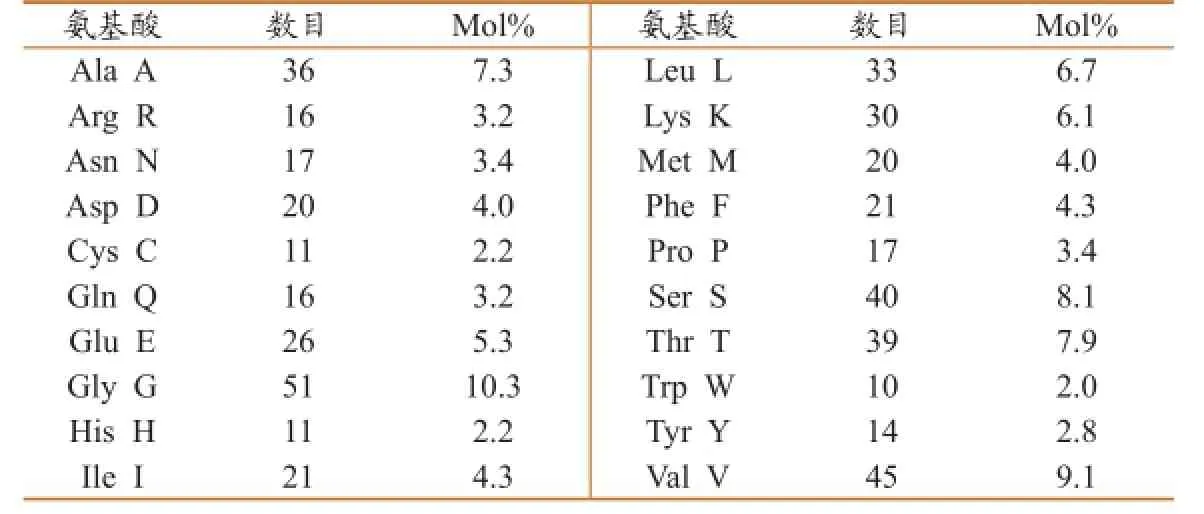
表1 DTMUV E基因序列推导肽链的氨基酸组分分析

图4 DTMUV E蛋白氨基酸序列的CDD分析结果

图5 Neural Networks (NN)对DTMUV E氨基酸序列推导肽链的信号肽预测结果
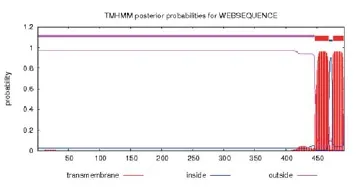
图6 DTMUV E蛋白序列跨膜区预测结果
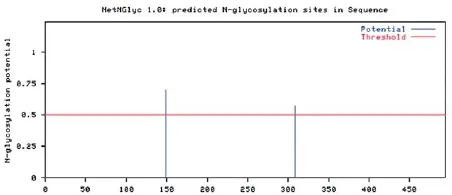
图7 DTMUV E氨基酸序列推导肽链的N-糖基化预测结果
2.2.3E蛋白信号肽预测结果。利用丹麦科技大学生物序列分析中心(CBS)网站在线程序 Signal P4.1对DTMUV E蛋白的氨基酸序列作信号肽预测,结果如图5。由图可见,DTMUV E蛋白序列中所有位点预测到的C值、S值或Y值的最大值都小于临界值,说明该蛋白没有信号肽切割位点,不含信号肽,成熟蛋白即为494aa。
2.2.4跨膜区预测结果。利用丹麦技术大学生物序列分析中心 (CBS)网站在线软件TMHMM 2.0预测DTMUV E蛋白跨膜区,结果见图6。跨膜区的预测分析结果表明该多肽链可能在451-468和475-492aa区域含有跨膜区,而且是膜外蛋白的可能性较大,这与其是衣壳蛋白也相符。
2.2.5E蛋白翻译后修饰预测
2.2.5.1糖基化位点预测。运用在线程序NetNGlyc1.0 对DTMUV E蛋白可能的N-糖基化位点进行预测,
结果表明在该序列中在149和309两个氨基酸位点上存在潜在的N-糖基化位点。
2.2.5.2磷酸化位点预测。运用在线程序NetPhos2.0预测DTMUV E蛋白磷酸化位点,预测结果见图8。结果显示当阈值为0.5时,共有26个潜在的磷酸化位点,其中含有15个丝氨酸磷酸化位点,7个苏氨酸磷酸化位点,4个酪氨酸磷酸化位点。
2.2.6DTMUV E蛋白的疏水性分析。利用在线 ProtScale 程序中的Wilson 疏水性分析法对DTMUV E蛋白质疏水性进行分析,结果如图 9。分析结果显示DTMUV E蛋白第461-467和484-490位氨基酸具有最强的疏水性。整个蛋白质疏水性最大值是4.689,最小值为-1.000,与疏水区相比,亲水区占据该蛋白质多肽链的区域大于疏水区域,表明该蛋白可能为亲水蛋白,这与其是一种衣壳蛋白相符。
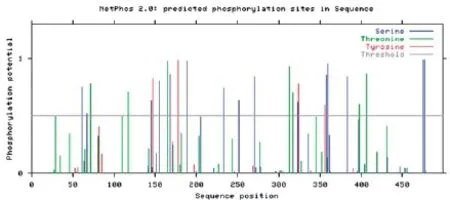
图8 DTMUV E氨基酸序列推导肽链的磷酸化位点的预测结果

图9 DTMUV E基因推导肽链的疏水性预测

图 10 DTMUV E基因推导蛋白质的二级结构预测

图11 DTMUV-E的三级结构预测
2.2.7DTMUV E蛋白的三级结构分析。利用http://www.rcsb.org/pdb/ explore/自动蛋白同源建模数据库进行蛋白全序列二级结构预测和3D结构预测,结果找到与登革热病毒E基因编码的在281-775区段相似的衣壳糖蛋白3D结构模型(图10),其三维构象(图11)主要包含α-螺旋和β-转角(Turn)。
2.3E基因的密码子偏爱性分析
应用EMBOSS在线软件CHIPS程序进行密码子使用频率统计,已知高表达基因其密码子偏爱程度大,从而Nc值较小;低表达基因含有较多种类的稀有密码子,Nc值则较大,所以,当前普遍通过比较Nc来确定内源基因表达量的相对高低。分析结果表明,DTMUV E的Nc值为55.037,表明该基因为相对低表达基因,含有较多种类的稀有密码子。
2.4DTMUV E基因的密码子与大肠杆菌、酵母及人的密码子偏爱性比较
根据EMBOSS软件CUSP程序分析比较其与大肠杆菌、酵母和人的三种表达系统的相似度。如表2所示,DTMUV-E与大肠杆菌等3种生物密码子使用频率比值(DTMUV-E /E.coli、DTMUV-E / Yeast、DTMUV-E /Human)在0.5~2之间的表示对该密码子的偏爱性较为接近,反之则差别较大(在小于0.5,大于2.0之间的频率比值采用下划线标志)。从表3的结果表明,DTMUV-E与大肠杆菌等3种密码子使用频率差值较大的在大肠杆菌有18个,酵母有16个,人有15个。由此可见,DTMUV-E的密码子使用偏爱性模式与差异并不大,若采用原核表达系统需注意密码子的优化使用,或采用截断表达方式更有利于DTMUV-E基因编码蛋白的外源表达。

表2 DTMUV-E与大肠杆菌、酵母及人的密码子偏爱性比较
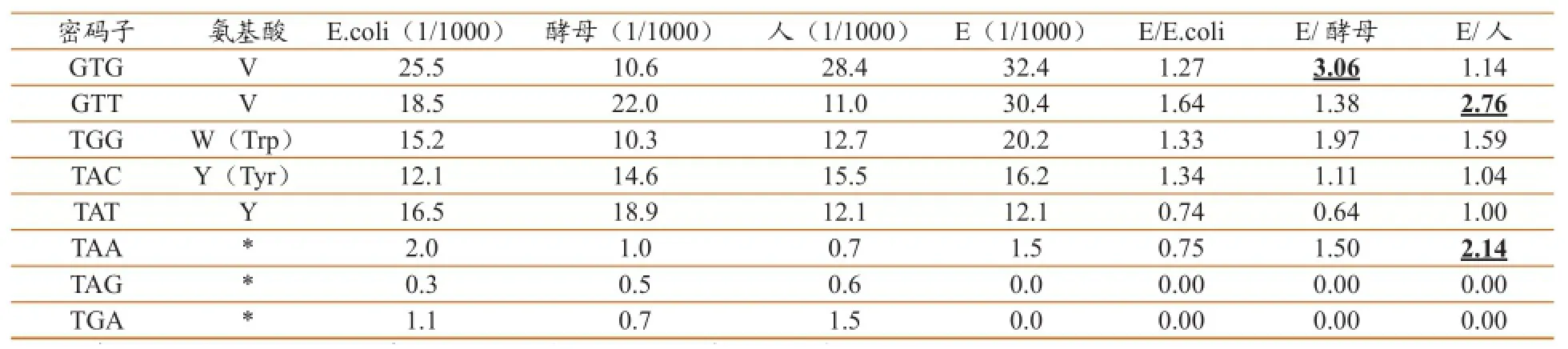
(续前)
3 讨论
3.1DTMUV-E基因的分子特性分析
本研究通过构建的DTMUV QD株基因文库重组质粒的DNA测序信息,结合NCBI的ORF Finder和BLAST工具分析得到了编码该病毒E蛋白基因的ORF,分子特性分析表明:该基因大小为1 488 bp,编码499个氨基酸,Clustal V工具预测表明该蛋白是类黄病毒衣壳蛋白家族成员之一。
根据E基因序列分析结果得知DTMUV与两个序列的相似性高达99%,核酸序列同源性比较结果显示该基因与坦布苏病毒属成员有较高的同源性。通常认为,蛋白水平之间达到25%的一致性就可提示其功能的相似性。三级结构搜索表明,本研究中预测的病毒株E蛋白与登革热病毒E基因编码的在281-775区段有着相似的衣壳糖蛋白3D结构模型(图2-图11),其三维构象主要包含α-螺旋和β-转角。这就提示它们在功能上可能具有一定相似性,同时也为我们下一步进行该编码蛋白的功能研究指明了方向。
3.2DTMUV-E密码子偏嗜性对表达的影响
选择合适的表达系统对于基因表达具有重要意义。密码子是核酸携带信息和蛋白质携带信息间对应的基本原则,是生物体内信息传递的基本环节。关于DTMUV在密码子使用上的偏爱性问题,目前国内外少有报道。本文对DTMUV-E编码基因的密码子偏爱性进行了分析,可以看出该蛋白中编码相同氨基酸密码子的偏爱性存在较大的差异。将分析结果通过与大肠肝菌、酵母和人的密码子偏爱性做进一步比较,可以看出DTMUV的密码子使用偏爱性模式与其差异并不大,若采用原核表达系统需注意密码子的优化使用,或采用截断表达方式更有利于DTMUV-E基因编码蛋白的外源表达。当然,外源基因的表达受到多种因素的共同作用,如要表达蛋白的种类、是否是毒素、是否需糖基化或其他修饰、是否需大量表达、如何纯化和蛋白生产成本等,而密码子用法的影响只是其中之一。通过密码子偏爱性分析不仅可为基因表达选择合适的表达系统,同时也可以为改造密码子进而提高基因的表达量提供依据。
3.3关于生物信息学及新基因的发现
大量DNA数据的存在有助于发现新的基因,由于实验测定的编码区域并不一定完整,必须结合计算找到并证实所有的外显子。就目前的分子生物学技术现状而言,实验测定DNA序列比测定蛋白质序列容易,因此可以通过实验测定一段基因的序列并由此推导蛋白质的氨基酸序列。在测出一段未知DNA序列后,往往要做的首要工作就是对序列数据库做一次相似性搜索,以确定获得的序列是否是一个新的未知序列或者数据库中是否已经存在与该序列同源的序列,从而发现序列的生物学意义。
[1]Su J,Li S,Hu X,et al. Duck egg-drop syndrome caused by BYD virus,a new Tembusu-related flavivirus[J]. PLoS One,2011,6(3):e18106.
[2]Yan P,Zhao Y,Zhang X,et al. An infectious disease of ducks caused by a newly emerged Tembusu virus strain in mainland China[J]. Virology,2011,417(1):1-8.
[3]曹贞贞,张存,黄瑜. 鸭出血性卵巢炎的初步研究[J]. 中国兽医杂志,2010,46(12):3-6.
[4]李译君. 鸭坦布苏病毒的分离鉴定及生物学特性研究[J].中国家禽,2011,33(17):34-35.
[5]马秀丽,于可响,高凤. 鸭黄病毒BZ株的生物学特性研究[J]. 中国家禽,2011,33(21):12-14.
[6]李玉峰. 一种从鸭新分离的黄病毒研究初报[J]. 畜牧兽医学报,2011,42(6):885-891.
[7]Emanuelsson O,Brunak S,von Heijne G,et al. Locating proteins in the cell using Target P,Signal P and related tools[J]. Nat Protoc,2007,2(4):953-971.
[8]Lambert C,Leonard N,De Bolle X,et al. ESyPred3D: Prediction of proteins 3D structures[J]. Bioinformatics,2002,18(9):1250-1256.
[9]Mullan L J,Bleasby A J. Short EMBOSS User Guide. European Molecular Biology Open Software Suite[J]. Brief Bioinform,2002,3(1):92-94.
[10]Zhao L C,Cheng A C,Wang M S,et al. Characterization of codon usage bias in the dUTPase gene of duck enteritis virus[J]. Progress in Natural Science,2008,18 (9)):1069-1076.
[11]Wright F. The 'effective number of codons' used in a gene[J]. Gene,1990,87(1):23-29.
(责任编辑:胡藕祥)
The Molecular Property Analysis of Duck Tembusu Virus E Gene
Sun Tao1,Wang Chao2,Deng Mingjun1,Wang Qun1,Zheng Xiaolong1,Xu Biao1
(1.Shandong Entry-exit Inspection and Quarantine Bureau,Qingdao,Shandong 266002;2. Tai’an Entry-exit Inspection and Quarantine Bureau,Tai’an,Shangdong 250014)
To provide the reference for the selection of antigen domain and gene expression host system of duck Tembusu virus,one recombinant plasmid comprising an ORF about 1488 bp was sequenced from DTMUV QD genebank combing NCBI ORF Finder and BLAST analysis. By analysis of NCBI BLASTP and DNAstar evolutionary tree,the ORF encoded protein have high similarity with the E gene of Flaviviriade virus class. Then the conservative structure domain was determined by Conserved Domain tools,signal peptide was predicted using Signal P4.1 and the trans-membrane region was predicted by TMHMM 2.0. Applying NetNGlyc1.0 and NetPhos2.0,ProtScale online,glycosylation sites,phosphorylation sites,hydrophobicity were analyzed. Finally,the fraction and using frequency of different codes in same amino acids were investigated by EMBOSS,and 3D structure was predicted by protein homology modeling analysis. Results showed:the E protein in this study have similar function with other Flaviviriade virus class capsid protein,no signal peptide cutting locus was discovered,and contained trans-membrane region in 451-468 and 475-492 AA area. And the E gene in this study which containing many kinds of rare codon,expression level was relatively low,codon usage frequency is close to with people. The study may provides the basis of molecular biology and contribute to in vitro expression selecting in further research.
duck tembusu virus;biological informatics;structure prediction
S852.65
A
1005-944X(2015)12-0059-08
徐 彪
猜你喜欢
上海金属(2021年6期)2021-12-02
福建农业学报(2021年6期)2021-08-18
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
教学考试(高考生物)(2020年4期)2020-11-18
生物学通报(2020年11期)2020-10-22
发明与创新·中学生(2019年6期)2019-06-26
中国洗涤用品工业(2019年4期)2019-05-11
电脑知识与技术(2018年19期)2018-11-01
中成药(2017年3期)2017-05-17
