
红曲取样方法再验证混合均一度偏差处理
2015-10-26 04:50北京北大维信生物科技有限公司100094王晓峰孙玉凤邓翀
首都食品与医药 2015年16期
北京北大维信生物科技有限公司(100094)王晓峰 孙玉凤 邓翀
我厂现红曲生产工艺的批量由3500kg变更至4000kg,原红曲取样验证方案适用于红曲生产工艺批量为3500kg,其所得结论“在生产过程中,出现容器改变、工艺控制参数改变或批量改变需要进行再验证”。现通过再验证,确认按照取样操作规程所取红曲批量为4000kg的红曲样品的洛伐他汀含量,能够正确反映整批的待出料红曲有效成分含量。评价取样方法的有效性、代表性和科学性,为证明取样方法的真实有效性提供基本依据。
红曲取样方法、取样量按《中国药典》(2010年版)第一部执行。经过风险评估,认为:取样量根据《药典》规定公式计算随批量扩大,不存在风险;红曲为分房间、分车、分瓶贮存,取样方法采用对角线交叉取样的方法,此方法已经过3500kg的取样验证,取样量扩大后,首先计算好取样瓶数,再根据房间的布局合理选择每车每列的取样瓶数,可以保证取样的代表性,不存在风险;原用于取样时样品混合的槽型混合机(设备型号CH100)不能满足现有样品混合量而采用新的取样用槽型混合机(设备型号CH200),因而存在槽型混合机不能充分混合均匀红曲的风险,和从槽型混合机中取出的红曲样品不能够正确反映整批的待出料红曲有效成分(洛伐他汀)含量的风险。
因此,风险评估得出的验证项目为:应确认所取红曲样品,在取样用槽型混合机中可以充分混合均匀,从槽型混合机中取出的红曲样品能够正确反映整批的待出料红曲有效成分(洛伐他汀)含量。通过分析验证过程中样品混合均一度测定数据(洛伐他汀含量相对标准偏差),最终验证我厂红曲取样方法在取样量扩大后,取样用槽型混合机能保证样品的混合均一度,能保证样品的代表性。
本验证过程均按照我公司验证管理规程执行,以下就验证实施过程、数据收集分析、验证结论等方面逐一阐述。
1 验证前准备
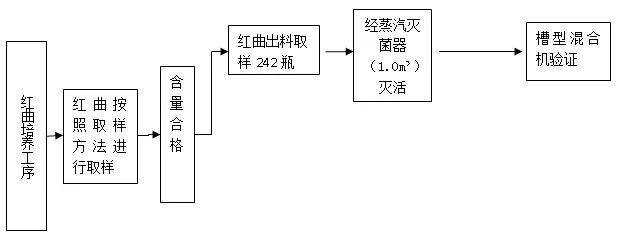
附图1 红曲取样流程图
1.1 人员 所有参加红曲取样方法再验证的相关操作人员、验证管理人员和QA生产监控员,在验证开始前应进行《红曲取样方法再验证方案》的培训,培训记录纳入培训档案。
1.2 关键仪器、设备确认检查 开始本次取样验证前,确认相关的仪器和设备确认已完成,且均符合要求,检查结果记录于《关键仪器、设备确认检查记录》。
关键设备槽型混合机(设备型号CH200,以下简称“槽型混合机”)容量200L,搅拌式混合方式,搅拌桨直径540mm,电机功率4KW,倒料角度105°,搅拌转速24r/min,重量550kg,出厂日期2003年12月,由江苏瑰宝集团有限公司生产,设备情况良好,运行正常,未进行大修,已经过容量核算和现场试验证明能满足4000kg红曲湿料样品的混合。
2 验证实施
2.1 取样批次和取样量
2.1.1 取样量 因红曲批量由3500kg变更为4000kg,按照《红曲取样SOP》(根据《药典》制定)进行最大量取样,取样量规定为:100<瓶数N≤1000时,按5%取样;瓶数N>1000瓶时,前1000瓶按照上述方法确定取样瓶数,超过部分按1%取样,取样瓶数计算若所得数值不是整数值,一律取比计算值大的第一个整数为准。(引自参考文献[1])而4000kg红曲最大可能瓶数为20133.3瓶。因此,总取样瓶数=50+(N-1000)×0.01=50+(20133.3-1000)×0.01=50+191.3=241.3(瓶)
向上取一位整数值,取样242瓶。
2.1.2 取样批次 选取红曲连续三批4000kg批量的红曲湿料,批号为H1、H2、H3。检测合格后,每批取242瓶,用蒸汽灭菌器(1.0m³)进行灭活处理用于验证。
2.2 取样方法 取样流程见附图1。
2.2.1 整批红曲的取样 采用对角线法按随机取样原则取样,直至取够规定样品瓶数。取出的红曲从发酵瓶转入槽型混合机混合。
2.2.2 槽型混合机内的取样 取样部位的选定参照《药品GMP指南》口服固体制剂关于混合设备混合均一度取样的规定:“至少应在上、中、下三个水平位置进行多点取样”。(引自参考文献[2])
取样部位:CH200型槽型混合机的搅拌桨是半叶式对称型,以轴为中心进行旋转搅拌,其与中心轴不同水平面的仓内四角(中心轴平面上层2角,下层2角)及双叶的对称中心点为易聚积物料处、混合效果最差,为取样点。
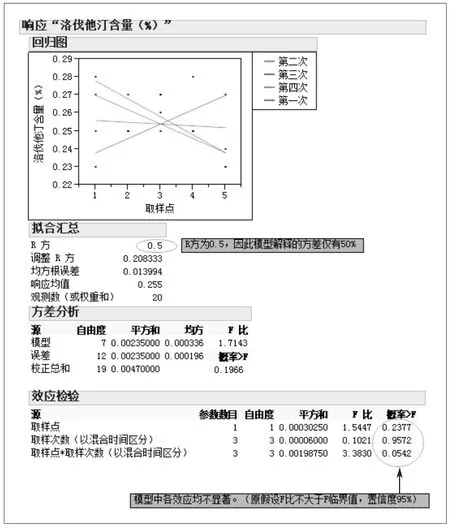
附图2 H3批洛伐他汀含量数据方差分析
2.3 取样 运行槽型混合机(速度24转/分钟),分别在混合的第1、2、3、4分钟进行取样。对仓内4角和中心点分别取样约60g,由QA生产监控员用取样勺在槽型混合机中心轴的中心点处取样,然后放入取样袋中记为取样点3;用取样勺紧贴料斗仓内四角,然后将物料取出,将取样勺中的样品放到取样袋中,分别记为取样点1、2、4、5;每个位点样品取3份(1份检验,2份备份),备份样品存放在QC理化冰箱内。QC检验人员按照红曲质量标准,用高效液相色谱法检测样品中的洛伐他汀的含量,将结果记录。
2.4 验证标准 槽型混合机每分钟混合后5个取样点之间液相含量相对标准偏差RSD应≤5.0%,最终以三批均能满足上述条件的混合时间为槽型混合机的样品混合时间。
3 数据收集与分析
3.1 三批次验证数据见附表1。
3.2 数据分析 H1批、H2批通过数据表明槽型混合机的混合3分钟能保证混合均匀;H3批数据超过预定标准,为偏差。
3.3 偏差分析
3.3.1 H3批洛伐他汀含量数据见附表2。
3.3.2 方差分析 使用JMP(版本10)对数据进行方差分析,α水平0.05,使用完全析因模型(模型效应:取样点,取样次数(以混合时间区分),取样点*取样次数(以混合时间区分),(方法出自参考文献[3])。结果及解释如下(见附图2)。
从分析可见,取样点、取样次数、取样点与取样次数交互作用的效应产生的变异对于误差项并不显著,因此,本次试验数据的变化可认为由其他误差项引起。
3.3.3 误差项分析 取样方法验证过程中,导致数据变异的因素,应为:样品均匀度、检验精密度,而《红曲取样方法再验证方案》中规定的“5个取样点之间液相含量相对偏差RSD应≤5.0%”,仅考虑了混合不均匀造成的变异。
检验精密度为1个样品6次检验相对偏差RSD应≤3.0%,结论来自《LC-20A高效液相色谱仪再确认报告》。所以,应同时考虑混合不均匀造成的变异和检验精密度造成的变异。
3.4 偏差处理
3.4.1 偏差处理原理 根据中心极限定理,从正态分布的总体取若干样品,亦呈正态分布;以样本标准偏差代替总体分布的标准偏差,以样本均值、样本标准差形成新的统计量t服从t分布。
利用t分布的覆盖因子k表(t分布的双侧分位数表),以样本量n的大小不同情况下对样本平均值的预测区间进行估计。本案例中取样是从被混合的产品这一正态总体中进行抽取的,洛伐他汀含量检验是对同一样品多次检验且在《LC-20A高效液相色谱仪再确认报告》确认检验结果的正态性的,因此,样本均值的预测区间均可以使用t分布进行估计(方法自参考文献[4]):

附表1 槽型混合机内5个取样点之间洛伐他汀含量相对标准偏差(RSD)

附表2 H3批洛伐他汀含量数据

附表3 液相含量相对偏差RSD标准的修改
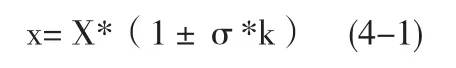
x为样品检测值;X为样品平均值;σ为样本相对标准偏差;k为n个样品的t分布的覆盖因子(90%置信度)
《药典》2010年版附录X E含量均匀度检查法使用90%置信度估计含量均匀的置信区间,因此使用90%置信度。(引自参考文献[1])
若样品均匀度造成的变异数据的t分布为RSD1,检验精密度造成的变异数据的t分布为RSD2,根据方差性质,此2总体合成的总体中样本的相对标准偏差σ为:
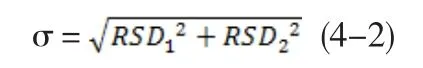
3.4.2 处理过程 红曲取样方法再验证混合均一度样品的液相含量相对偏差RSD标准的修改如附表3所列。
《药品GMP指南》口服固体制剂规定:“混合均一度应该控制在85%~115%或更严格的工艺指标,相对标准偏差不应高于7.8%” (引自参考文献[2]),因此x∈ [85%X,115%X]。由(4-1),即X*(1±2.13σ)∈ [85%X,115%X],计算得σ≤7.04%,可见σ同时满足《药品GMP指南》“相对标准偏差不应高于7.8%”的规定。
故在验证混合均一度时,应同时考虑混合不均匀造成的变异、检验精密度造成的变异,并宜根据“混合均一度应该控制在85%~115%或更严格的工艺指标,相对标准偏差不应高于7.8%”的规定制定验证数据总体的相对标准偏差的标准。本次验证以不增加相对标准偏差的原则进行调整为:±7.0%,即相对标准偏差标准定为RSD≤7.0%。
3.4.3 偏差处理结论 偏差经分析并做调整样品相对标准偏差标准的处理后,偏差可接受。根据RSD≤7.0%的标准,H1、H2、 H3数据表明槽型混合机(设备型号CH200)混合2分钟均能保证混合均匀。为保证混合时间余量,取混合时间为3分钟。
4 结论
通过对红曲取样方法进行再验证,验证结果无遗漏,已对偏差进行合理说明;不需要进一步补充试验。验证实施过程中对验证方案有1处修改:“槽型混合机每分钟混合后5个取样点之间液相含量相对标准偏差RSD应≤5.0%”,改为“槽型混合机每分钟混合后5个取样点之间液相含量相对标准偏差RSD应≤7.0%”。最终证实:验证结果真实有效,取样时可以使用槽型混合机(设备型号CH200)混合,混合速度24转/分钟,混合时间3分钟;红曲(4000kg)的取样方法取得的样品,能够代表整批红曲的真实含量。
根据验证结果,将《红曲取样操作程序》变更如下:由“槽型混合机(设备型号CH100,设备编码XXXX)混合3分钟”变更为“槽型混合机(设备型号CH200,设备编码XXXX)混合3分钟”。操作程序变更后,对QA取样人员进行培训,以保证验证结论的执行。
猜你喜欢
现代食品(2022年19期)2022-11-21
科学技术创新(2021年19期)2021-07-16
中老年保健(2020年5期)2020-12-11
食品与发酵工业(2019年5期)2019-03-28
食品与发酵工业(2019年2期)2019-02-15
食品与发酵工业(2018年12期)2019-01-14
中国药剂学杂志(网络版)(2019年6期)2019-01-05
中央民族大学学报(自然科学版)(2018年1期)2018-10-22
科技创新与应用(2017年1期)2017-05-11
科技与创新(2016年10期)2016-05-28
