
Ⅰ型神经纤维瘤病合并双侧腹膜后恶性肿瘤1例报告并文献复习
2015-06-24 14:34张立进张士伟赵晓智刘光香孟凡青郭宏骞
现代泌尿外科杂志 2015年8期
张立进,张士伟 ,赵晓智,刘光香,孟凡青,郭宏骞
(南京大学医学院附属鼓楼医院:1.泌尿外科;2.病理科,江苏南京 210008)
·临床研究·
Ⅰ型神经纤维瘤病合并双侧腹膜后恶性肿瘤1例报告并文献复习
张立进1,张士伟1,赵晓智1,刘光香1,孟凡青2,郭宏骞1
(南京大学医学院附属鼓楼医院:1.泌尿外科;2.病理科,江苏南京 210008)
目的 探讨Ⅰ型神经纤维瘤病合并恶性外周神经鞘瘤与恶性嗜铬细胞瘤的临床、病理学特征,提高对此类疾病的诊疗认知水平。方法 回顾性分析我院收治的1例Ⅰ型神经纤维瘤病合并不同性质恶性肿瘤的临床资料,总结归纳其临床及病理特征,并结合相关文献,分析此类疾病的相关特征及治疗预后。 结果 本例患者成功施行双侧腹膜后恶性肿瘤切除术,术后左侧提示恶性外周神经鞘瘤,免疫组化示:S -100(+),Ki-67(+),SMA(-),HMB45(-),右侧示恶性嗜铬细胞瘤,免疫组化示:Syn(+),CgA(+),Ki-67(+),皮肤结节病理提示神经纤维瘤。术后1月后给予IVP化疗方案化疗6个疗程,随访半年余,未见肿瘤复发或转移。结论 Ⅰ型神经纤维瘤病同时合并恶性外周神经鞘瘤与恶性嗜铬细胞瘤为临床十分罕见病例,恶性程度相对较高,预后较差。治疗以手术治疗为主,辅以化疗等相关治疗具有一定疗效,但具体疗效仍需相关临床随访观察。
Ⅰ型神经纤维瘤病;恶性外周神经鞘瘤;恶性嗜铬细胞瘤
神经纤维瘤病(neurofibromatosis,NF)又称多发性神经纤维瘤,其发病率约为1/2 500~1/3 000,无性别和种族差异[1]。而恶性外周神经鞘瘤(malignant periphelal nerve sheath tumors,MPNSTs)为来源于周围神经系统或神经外软组织肿瘤,可发生于身体任何部位,发病率约占NF的3%,且为NF患者的主要死因[2]。恶性嗜铬细胞瘤(malignant pheochromocytoma)约占肾上腺嗜铬细胞瘤的15%,但隐匿性恶性嗜铬细胞瘤在临床上相当罕见。国内外文献已有神经纤维瘤Ⅰ型合并恶性嗜铬细胞瘤或者恶性神经鞘瘤的相关报道[2-3],但NF同时合并恶性外周神经鞘瘤与无功能性恶性嗜铬细胞瘤却尚未有人报道。作为首次报道的相关病例,现结合相关已有文献及我科诊治经验,汇总报道如下。
1 病例报告
患者男性,44岁,因“上腹部间歇性疼痛1月余”入院。患者1月前无明显诱因出现上腹部疼痛,为钝痛,持续数分钟后可缓解,发作时无冷汗、心悸、头晕等症状。当地医院就诊,查MRI示两侧腹膜后占位(右侧肾上腺区较大),结合腹壁多发结节,考虑神经纤维瘤病。后患者转至我科进一步治疗。患者既往体健,自诉20岁时胸腹部开始出现细小结节,近年长大、增多。否认家族遗传史,无高血压等病史。入院检查:血压141/74 mmHg,全身可见散在分布的大小不一的皮下结节,最大约3.2 cm×1.3 cm,表面光滑,质软,无压痛,边界清楚,颜色较周边皮肤稍深,结节间弥散分布淡咖啡色斑(图1)。左右季肋区分别可扪及包块,其中右侧较大,质中,无压痛,移动性浊音(-)。专科查体无明显异常。辅检尿儿茶酚胺:E 3.64 μg/24 h;NE 69.97 μg/24 h;多巴胺 57.90 μg/24 h、血皮质(8∶00、16∶00、24∶00):187.00、93.50、37.50 nmol/L;血ACTH(8∶00、16∶00、24∶00):3.22、3.13、2.02 pmol/L、血肾素(卧位、立位):1.17、6.18 ng/(mL·h);血醛固酮(卧位、立位):99.94、101.83 pg/mL;血钾:4.42 mmol/L、钠:135.7 mmol/L;复查CT示:双侧腹膜后占位,双肾受压移位,增强后呈不均匀强化(图2)。

图2 双侧腹膜后肿瘤CT影像学表现
A:左侧恶性神经鞘瘤,包膜尚完整(黑色箭头所示),但肿瘤基底与左侧腰大肌分界不清,呈不均匀强化。右侧隐匿性恶性嗜铬细胞瘤,其内密度不均,局部可见坏死液化(白色箭头所示); B:CT三维重建可见双侧腹膜后恶性肿瘤丰富血供。
患者完善术前相关检查,排除手术禁忌,行经腹双侧腹膜后肿瘤切除术。术中探查见:左侧肿瘤位于左肾动静脉下方,肿瘤假包膜完整,周边紧靠左肾及腔静脉,但肿瘤基底与腰大肌粘连紧密,无法连同假包膜取出。遂充分游离肿瘤周边脏器并深层次游离肿瘤假包膜,取出左侧肿瘤,约10 cm×8 cm×6 cm,术中快速病理示:梭形细胞肉瘤。右侧肿瘤较大,约16 cm×14 cm×10 cm,肿瘤包膜完整,表面血供丰富,与周围脏器界限清楚。术中处理右侧肿瘤时,患者血压波动较大,最高达250/143 mmHg,术中考虑可能为隐匿性嗜铬细胞瘤。快速分离取出肿瘤后,患者血压恢复正常。随机切取患者右下腹表皮结节,连同两侧肿瘤送病理检查。总手术时长4 h,失血量约600 mL。术后左侧肿瘤病理示低分化梭形细胞肉瘤,结合免疫组化考虑恶性外周神经鞘瘤。肿瘤切面呈暗红色、灰白色相间,部分区域出血坏死,免疫组化示:CD117(-),CD34散在(+),SMA(-),Desmin(-),S -100散在(+),HMB45(-),Bcl2(-),CK(-),Ki-67最密接处约30%(+)(图3)。右侧恶性嗜铬细胞瘤,肿瘤切面呈囊实性,囊内含多量血性液体,免疫组化示:Syn(+++),CgA(+++),A103(-),S -100散在(+),Inhibina散在(+),Ki-67<5%(+)(图4)。皮肤结节病理示神经纤维瘤,切面呈灰白色,免疫组化示:S -100(+),EMA弱(+)。诊断:①Ⅰ型神经纤维瘤病;②恶性外周神经鞘瘤;③隐匿性恶性嗜铬细胞瘤。患者术后恢复可,无头晕、心悸等不适主诉,血压平稳,术后血压维持在105~127/97~83 mmHg。患者术后未予激素补充等特殊治疗,1个月后根据病理给予患者IVP方案化疗(异环磷酰胺 2 g, 第1、2、3天,并于给药后第0、4、8 小时给予美司钠400 mg保护泌尿系;依托泊苷 100 mg,第1、2、3天;顺铂 40 mg,第1、2、3天)化疗,3周为一疗程,总疗程4~6次。化疗过程中给予充分水化,并提升白细胞、止吐等对症支持治疗,同时予善龙(醋酸奥曲肽微球)20 mg皮下注射,1次/月。患者术后已行6个周期化疗,随访至今,未见肿瘤局复发或远处转移。
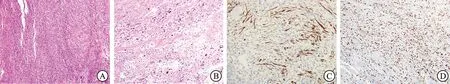
图3 恶性外周神经鞘瘤HE及免疫组化染色
A:恶性外周神经鞘瘤典型栅栏样神经纤维结构(HE,×100);B:明显核异性细胞(HE,×100);C: S -100免疫组化染色(×200)示肿瘤组织内典型梭形细胞; D:Ki-67染色阳性,阳性率大于30%,肿瘤细胞恶性程度指数活跃(×100)。
A:恶性嗜铬细胞瘤胞质嗜双色性,局部组织细胞坏死明显(HE,×100);B: CgAZ在肿瘤组织内强阳性表达,阳性表达主要位于胞质(×100);C:肿瘤组织内S -100染色染色呈阳性反应(×200);D:肿瘤组织内Syn高表达,阳性表达主要位于胞质,部分位于包膜(×100)。
2 讨 论
NF属常染色体显性遗传综合征,由VIRCHOW于1847年首次报道。按照美国国家卫生研究院(National Institutes of Health,NIH)制定的NF诊断和分型标准,本病可分为NF-Ⅰ型与NF-Ⅱ型[4]。临床上一般习惯分为:①中枢型:以中枢神经系统的胶质瘤、脑膜瘤等肿瘤为主;②内脏型:以内脏或自主神经系统为主的肿瘤,如神经纤维瘤、恶性神经鞘瘤等;③顿挫型:主要表现为外周皮肤色素沉着;④周围型:外周多发性神经纤维瘤和丛状神经纤维瘤,并伴有皮肤咖啡斑,也可伴有其他先天性畸形。本例患者按NIH分型属于NF-Ⅰ型,按临床分型则属于内脏与周围混合型,为十分罕见类型NF。
恶性外周神经鞘瘤又称恶性神经鞘瘤,是一种来源于外周神经的低分化梭形细胞肉瘤,年发病率约为1/10万,占软组织肉瘤的5%[2]。恶性神经鞘瘤可发生于身体任何部位,以四肢和躯干多见,但原发于腹膜后者罕见[5]。腹膜后恶性神经鞘瘤CT可表现为孤立或弥漫性肿块,大小不一,其内成分丰富,密度不均。增强时,肿瘤实质可呈斑块状、岛屿状强化,且常压迫、侵犯周围组织[6]。本例瘤体CT表现并不十分明显,但可见腰大肌明显受侵犯,且CT三维重建可见肿瘤侵及腰椎旁动脉(图2B)。MPNST可通过病理确诊,光镜下常为纤维肉瘤生长类型,肿瘤细胞以梭形细胞为主,伴有核扭曲等异常核分裂象。但由于软组织肉瘤细胞形态和组织结构的多样性,常规HE染色切片常难以鉴别。免疫组织化学是重要的辅助诊断和鉴别诊断的手段,S -100、Leu-7、髓鞘碱性蛋白或超微结构为常用检测指标。文献报道S -100(+),cytokeratin(-),SMA(-),HMB45(-)对于诊断恶性外周神经鞘瘤具有高度敏感性[7]。本例S -100阳性,Ki-67>30%,SMA(-),HMB45(-),结合HE染色,诊断为恶性外周神经鞘瘤。
嗜铬细胞瘤为一种少见肿瘤,其发病率约为十万分之一,而恶性嗜铬细胞瘤更不足其10%[3]。静息性恶性嗜铬细胞瘤作为恶性嗜铬细胞瘤的一种,临床上一般分为隐匿功能性嗜铬细胞瘤与无功能性嗜铬细胞瘤。本例患者既往无高血压病史,入院后血压监测亦无明显异常,手术过程中因刺激瘤体造成术中血压剧烈波动,结合临床特征,诊断为隐匿型恶性嗜铬细胞瘤。与典型嗜铬细胞瘤相比,隐匿性恶性嗜铬细胞瘤临床表现及各项生化检查均不典型,且与良性嗜铬细胞瘤病理形态相似,给疾病的早期诊断带来许多困难。通常认为肾上腺肿瘤侵及邻近脏器或在远处出现转移灶,即可诊断为恶性嗜铬细胞瘤,但肿瘤发生转移时间可能过长,因此在早期需要寻找能够区别良恶性嗜铬细胞瘤的指标。刘彤华等[8]人研究发现,Ki-67>3%,出现不典型分裂或核坏死,则高度考虑恶性嗜铬细胞瘤。而临床上一般以肿瘤直径>5 cm,重量>80 g,肿瘤侵犯血管或者包膜,出现融合性坏死来虑恶性嗜铬细胞瘤。虽然到目前为止除Ki-67指标外,其他肿瘤标志物对于鉴别良恶性嗜铬细胞瘤均无特异性[9]。本例Ki-67虽小于5%,但大于文献报道的3%标准,且同时结合临床判断标准,可以诊断为隐匿性恶性嗜铬细胞瘤。
NF1为NF-Ⅰ型的致病基因,而为作为一种肿瘤抑制基因,其编码的神经纤维瘤蛋白(neurofibromin)可在神经元、施旺细胞等神经细胞中高表达,具有肿瘤生长抑制功能。研究发现,Neurofibromin 能够激活GTP酶,催化Ras蛋白水解,使原癌基因Ras基因失活。而NF-Ⅰ患者的基因突变使得原癌基因Ras通路激活,且RF1基因突变常发生于胚胎早期,故可造成全身多系统受损。NF1的主要临床表现包括全身皮肤多发咖啡色斑、弥散的皮肤神经纤维瘤,其中咖啡斑为本病的一个重要体征。尽管NF1为良性肿瘤,但患有NF1的患者得其他良性或恶性肿瘤几率明显提高,且研究发现NF1易并发其他器官或系统等病变,如骨骼畸形、中枢神经系统肿瘤、嗜铬细胞瘤等[10]。本文报道的NF1同时合并MPNSTs与恶性嗜铬细胞瘤,为国内外罕见病例,至今尚无相关文献报道。恶性外周神经鞘瘤可为原发恶性肿瘤,也可也可由外周型神经纤维瘤等良性肿瘤恶变而来。而恶性嗜铬细胞瘤通常以散发为主,但30%与遗传性肿瘤有关,研究发现,NF1与恶性神经鞘瘤在成人中的关联性可达11.5%[3]。本例患者虽无相关NF1家族史,但综合相关文献资料与患者临床表现,我们认为左侧MPNSTs与右侧恶性嗜铬细胞瘤是并发于NF1系统性疾病所致。
目前针对NF1的治疗尚无明确的治疗方式,治疗的目标主要集中在预防相应的并发症上,治疗方式包括手术治疗、放化疗、基因治疗等。综合相关文献分析,彻底的手术切除治疗无论是对于恶性嗜铬细胞瘤还是恶性神经鞘瘤都是首选,但是相比单纯手术而言,手术联合放化疗效果更好。CARLI等[11]研究发现,手术联合术后放疗能显著延长患者的生存期及减少术后肿瘤的复发。虽然到目前为止针对化疗在MPNSTs的作用尚有不同观点,但临床研究表明多柔比星联合异环磷酰胺能够降低恶性神经鞘瘤复发率,且对于那些较大肿瘤,术前化疗能够使其明显缩小,为手术切除创造最佳条件[12]。恶性嗜铬细胞瘤一般预后较差,文献报道非手术治疗患者5年生存期小于50%,而手术治疗患者生存期可显著延长,部分患者可长期存活。临床上因瘤体较大或者与周围组织粘连紧密无法完整切除时,术后的辅助化疗具有重要意义,目前恶性嗜铬细胞瘤公认的化疗方案为CVD(环磷酰胺、长春新碱、达卡巴嗪)[13]。该患者病理结果显示右侧肿瘤Ki-67指数较左侧低,右侧肿瘤恶性度相比而言无左侧高,且从手术角度来说,右侧肿瘤切除较左侧彻底,因此结合相关文献报道,我们以异环磷酰胺为基础,结合铂类化疗药物治疗左侧MPNSTs,并予善龙治疗右侧恶性嗜铬细胞瘤。
综上所述,NF1合并恶性神经鞘瘤与恶性嗜铬细胞瘤为十分罕见疾病。本例患者两侧腹膜后恶性肿瘤较大,且两侧肿瘤性质、成分不一,对化疗敏感度也不尽相同。但从手术治疗方面来说,该患者手术效果取得满意效果,但该患者对于化疗的反应性及远期生存时间仍需要长期随访观察。
[1] HIRBE AC, GUTMANN DH. Neurofibromatosis type 1: a multidisciplinary approach to care[J]. Lancet Neurol,2014,13(8):834-843.
[2] POURTSIDIS A, DOGANIS D, BAKA M, et al. Malignant Peripheral Nerve Sheath Tumors in Children with Neurofibromatosis Type 1[J]. Case Rep Oncol Med, 2014,2014:1-6.
[3] GIOVANNONI I, CALLEA F, BOLDRINI R, et al. Malignant pheochromocytoma in a 16-year-old patient with neurofibromatosis type 1[J].J Ped pathol,2014,17(2):126-129.
[4] BOYD KP, GAO L, FENG R, et al. Phenotypic variability among cafe-au-lait macules in neurofibromatosis type 1[J]. J Ame Acad Dermatol,2010,63(3):440-447.
[5] WONG CS,CHU TY,TAM KF.Retroperitoneal schwannoma:a common tumour in an uncommon site[J].Hong Kong Med J,2010,16(1):66-68.
[6] 建军,王建华,曾蒙苏,等.腹膜后恶性神经鞘瘤的影像表现[J].中华放射学杂志,2009,43(4):432-434.
[7] ÖZDAL B,ÖZ M, KORKMAZ E, et al. Malignant peripheral nerve sheath tumor of the vulva, an unusual differential diagnosis for vulvar mass[J].Int J Surg Case Rep,2014,5(11):793-795.
[8] 刘彤华,陈原嫁,武莎菲,等.良性和恶性嗜铬细胞瘤的区别[J].中华病理学杂志,2004,33(3):199.
[9] STRONG VE, KENNEDY T, AHMADIE AH, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis[J]. Surgery,2008,143(6):759-768.
[10] ANGHILERI M, MICELI R, FIORE M, et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution[J]. Cancer,2006,107(5):1065-1074.
[11] CARLI M, FERRARI A, MATTKE A, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group[J]. J clin oncol: J Ame Soci clin onclo,2005,23(33):8422-8430.
[12] KROEP JR, OUALI M, GELDERBLOM H, et al. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma group study[J]. Ann oncol,2011,22(1):207-214.
[13] DEUTSCHBEIN T, FASSNACHT M, WEISMANN D, et al. Treatment of malignant phaeochromocytoma with a combination of cyclophosphamide, vincristine and dacarbazine: own experience and overview of the contemporary literature[J]. Clin endocrinol, 2015,82 (1): 84-90.
(编辑 王 玮)
Neurofibromatosis Ⅰ with bilateral malignant retroperitoneal tumor: a case report and literature review
ZHANG Li-jin1,ZHANG Shi-wei1,ZHAO Xiao-zhi1,LIU Guang-xiang1,MENG Fan-qing2,GUO Hong-qian1
(1. Department of Urology; 2. Department of Pathology, Nanjing Drum Tower Hospital, Affiliated Hospital, Medical School of Nanjing University, Nanjing 210008, China)
Objective To explore the clinical and pathological features of neurofibromatosis I combined with malignant peripheral nerve sheath tumor (MPNST) and malignant pheochromocytoma, as to improve the level of diagnosis and treatment of this disease. Methods Clinical data of 1 case of neurofibromatosis I with two different malignant tumors were retrospectively analyzed and relevant literature was reviewed. Results The operation was performed and bilateral retroperitoneal tumors were successfully removed. The pathological results showed that the left tumor was MPNST. Immunohistochemical staining showed: S -100(+), Ki-67(+), SMA(-), HMB45(-). The right tumor was malignant pheochromocytoma. Immunohistochemical staining showed: Syn(+), CgA(+), Ki-67(+). The skin nodules were proved to be neurofibroma. Six sessions of chemotherapy regimens of IVP were given to the patient a month later after the surgery. No tumor recurrence or metastasis was detected. Conclusions NeurofibromatosisⅠcombined with MPNST and malignant pheochromocytoma is a rare event. An appropriate surgery with adjuvant chemotherapy might be effective to some extent. But the prognosis of this disease is poor for it’s high degree of malignancy. More relevant studies are needed for further observation.
neurofibromatosis I; malignant peripheral nerve sheath tumor; malignant pheochromocytoma
2015-01-30
2015-05-09
郭宏骞,教授,主任医师,博士生导师.E-mail:dr.ghq@163.com
张立进(1990-),男(汉族),硕士在读.研究方向:泌尿生殖系统肿瘤.E-mail:stzlj913729553@163.com
R699.3
A
10.3969/j.issn.1009-8291.2015.08.013
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
透析与人工器官(2020年1期)2020-11-16
透析与人工器官(2020年1期)2020-11-16
临床肝胆病杂志(2020年9期)2020-09-28
中国临床医学影像杂志(2019年6期)2019-08-27
中国临床医学影像杂志(2019年6期)2019-08-27
中国临床医学影像杂志(2019年5期)2019-08-27
现代泌尿生殖肿瘤杂志(2018年5期)2018-11-29
现代泌尿外科杂志(2018年5期)2018-06-07
中国现代神经疾病杂志(2018年2期)2018-05-09
