
低氧预处理激活神经元细胞JAK/STAT并促进血管内皮生长因子受体-2的表达
2015-06-24 14:31韦可聪朱云中梁韡斌张高炼
中国神经免疫学和神经病学杂志 2015年1期
韦可聪 朱云中 梁韡斌 张高炼
低氧预处理激活神经元细胞JAK/STAT并促进血管内皮生长因子受体-2的表达
韦可聪 朱云中 梁韡斌 张高炼
目的 研究低氧预处理对神经元细胞Janus激酶(JAK)/信号转导和转录激活因子(STAT)信号通路以及血管内皮生长因子受体-2(VEGFR-2)表达的影响。方法 将培养的小鼠神经元细胞分为细胞对照组、细胞类缺血组和细胞低氧预处理组,细胞类缺血组细胞经95% NO2+5% CO2混合气体处理30 min,细胞低氧预处理组细胞于89%N2、l%O2和10%CO2环境中处理8次,每次20 min,2 d后进行类缺血处理。定量分析各组细胞中VEGFR-2、JAK及STAT蛋白磷酸化(p-STAT)的表达变化。细胞转染JAK 特异性小干扰RNA(siRNA),之后进行低氧预处理和类缺血处理,检测p-STAT和VEGFR-2的表达。另取45只BALB/c小鼠分为动物对照组(n=8)、动物对照+血管内皮生长因子(VEGF)组(n=7)、动物脑缺血组(n=8)、动物脑缺血+VEGF组(n=7)、动物低氧预处理组(n=8)和动物低氧预处理+VEGF组(n=7),分析各组海马神经元细胞p-STAT和VEGFR-2的表达,并检测细胞中含半胱氨酸的天冬氨酸蛋白水解酶(caspase 3)的活化情况。结果 与细胞对照组相比较,细胞类缺血组细胞中VEGFR-2 mRNA表达显著下调(P<0.05);与细胞类缺血组相比,细胞低氧预处理组细胞VEGFR-2 mRNA表达上调(P<0.05),并促进JAK mRNA和蛋白的表达(P<0.05)以及STAT蛋白的活化(P<0.05)。与非特异性转染组相比,JAK沉默组细胞p-STAT和VEGFR-2的表达显著降低(均P<0.05)。在小鼠体内,脑缺血降低p-STAT和VEGFR-2的表达(P<0.05),而低氧预处理则可有效逆转缺氧对p-STAT和VEGFR-2的抑制(P<0.05)。与动物脑缺血+VEGF组相比,动物低氧预处理+VEGF干预可显著抑制caspase3的活化(P<0.05)。结论 低氧预处理可以激活神经元细胞JAK/STAT信号通路并诱导VEGFR-2的表达,并增强VEGF的神经保护作用。
缺氧,脑;缺血预处理;细胞内信号肽和蛋白质类;Janus激酶类;JAK/STAT;血管内皮生长因子受体-2
脑缺血预处理可诱导机体产生缺血耐受[1],而低氧预处理能使许多器官对再次的缺血缺氧有保护作用,可以诱发机体的自我保护机制[2]。低氧预处理诱导缺血耐受与细胞内多种分子机制相关,其中包括诱导血管内皮细胞生长因子(vascularendothelialgrowthfactor,VEGF)合成[3-4]。血管内皮细胞生长因子受体-2(vascularendothelialgrowthfactorreceptor-2,VEGFR-2)是VEGF受体之一,研究发现VEGFR-2的表达与Janus激酶(Januskinase,JAK)/信号转导和转录激活因子(signaltransducersandactivatorsoftranscription,STAT)相关[5],然而目前尚不清楚低氧预处理诱导的神经保护是否与VEGFR-2表达及JAK/STAT激活有关。本研究利用低氧预处理培养的神经元和小鼠,观察小鼠海马神经元和培养神经元VEGFR-2及JAK/STAT的表达变化,旨在为阐明缺血耐受机制提供依据。
1 材料和方法
1.1 观察
1.1.1 神经元的原代培养:选取新生24h内的BALB/c小鼠,乙醇消毒后处死取脑,分离脑海马,用0.25%(质量浓度)的胰酶结合机械吹打的方法制备单细胞悬液,调节细胞水平至106/mL,种板。首次换液于种板24至48h进行,培养到第3~4天。加入阿糖胞苷(10μmol/L)抑制胶质细胞生长,纯化神经元。处理1~2d后全量换液,以后每周换液2~3次,每次半量换液。
1.1.2 实验动物:45只成年雄性BALB/c小鼠购自上海斯莱克实验动物有限公司,体质量20~35g,饲喂在(24±2)℃,(50±10)% 相对湿度条件下。实验动物的使用严格遵守本院伦理道德委员会制定的指导原则,并且尽量减轻实验动物所受的损伤和痛苦。
1.2 主要试剂 阿糖胞苷和MEM培养基购自Gibico公司,噻唑蓝(MTT)试剂购自美国Amresco公司,RNA-SolveReagent试剂盒购自Invitrogen,DyNAmoSYBRGreenqPCR试剂盒购自宝生物工程有限公司,兔抗鼠p-STAT抗体、procaspase3和cleavagecaspase3以及酶标羊抗兔二抗均购自英国Abcam。
1.3 方法
1.3.1 低氧预处理:将培养的神经元细胞分为3组,即细胞对照组、细胞类缺血组和细胞低氧预处理组。(1)细胞低氧预处理组:将神经元接种于24孔培养板中,定时将细胞移入2000cm3充有低氧气体(89%N2+l%O2+10%CO2,均为体积分数)的恒温密闭容器内(保持温度36℃),低氧条件下处理20min后立即取出,再恢复常氧培养。细胞每天定时接受20min低氧预处理,并反复处理8次,之后进行类缺血处理。(2)细胞对照组:在常氧环境中培养。(3)细胞类缺血组:直接进行类缺血处理:将神经元细胞移入37℃恒温密闭容器中,连续充以95%NO2+5%CO2(均为体积分数)混合气体,流速为20mL/min,持续30min。
1.3.2 小鼠脑缺血造模及分组:参考Koizumi等[6]与Longa等[7]方法,采用颈内动脉尼龙线(3-0)线栓法,建立小鼠脑缺血模型。术中保留自主呼吸,体温由肛温探头连接多功能监测仪(Spacelab,USA)监测,并用烤灯维持在37.0~37.5℃。所有造模小鼠均造模成功,未发生死亡现象。将45只小鼠随机分为动物对照组、动物脑缺血组、动物低氧预处理组,每组15只。动物对照组小鼠只分离中动脉但不夹闭。动物低氧预处理组小鼠先置于8%O2+92%N2(均为体积分数)的环境中处理2h,而后建立脑缺血模型。各组动物随机抽选7只接受VEGF治疗,经尾静脉注射5μg/mLVEGF盐溶液,共治疗7d,另8只小鼠处死用于检测p-STAT和VEGFR-2表达。
1.3.3MTT法检测细胞的生长情况:各组细胞经处理后弃去上清液,磷酸盐缓冲液(PBS)清洗,每孔加入MTT100μL(终浓度为0.15mg/mL),置于5%(体积分数)CO2培养箱内继续孵育4~6h,加入10%(质量浓度)十二烷基硫酸钠与50%(体积分数)异丙醇(100μL/孔)助溶。37℃放置过夜后于全自动酶标仪490nm波长处测定吸光度〔D(λ)〕值。实验重复3次,取平均数作为实验结果。
1.3.4siRNA的合成和转染:根据JAK的基因序列,设计含有SalⅠ 和XbaⅠ酶切位点的siRNA序列,序列1为:5′-GTCGACGTCTTGGTCTACAGGCTACGCTCTAGA-3′,序列2为:5′-GTCGACCAGTTACAAAGCCTGTCTGCCTCTAG-A-3′,引物通过SalⅠ-XbaⅠ限制性位点克隆到shRNA质粒pAVU6+27中,重组质粒序列由测序进行鉴定。阴性对照siRNA序列为:5′-GTCGACAGTTCACAACTAGCTCGTCCGTC-TAGA-3′,序列由上海生工合成。细胞接种到24孔培养板中,取5 μL LipofectamineTM2000稀释到250 μL减血清培养基MEM(minimum essential media)培养基中,轻轻混匀,室温孵育5 min;取8.0 μL siRNA稀释到上述含5 μL LipofectamineTM2000的250 μL减血清MEM培养基中,室温孵育20 min。取50 μL siRNA-Lipofectamine复合物加入到细胞培养孔中,轻轻摇动混匀,37 ℃,5% (体积分数)CO2条件下培养。转染24 h后,荧光显微镜检测转染效率,转染效率为17.5%。
1.3.5 荧光定量PCR方法检测mRNA表达水平:利用RNA-Solve Reagent 试剂盒提取培养神经元细胞总RNA,反转录合成cDNA。以cDNA为模板合成VEGFR-2、JAK基因片断扩增,扩增所用引物如表1所示。扩增产物电泳后染色、观察。基因表达的测定采用DNA Engine OpticonTM2荧光检测系统和DyNAmo SYBR Green qPCR 试剂盒。目的基因和β-actin 基因的拷贝数分别根据产生的Ct值从各自的标准曲线获得。取3次重复的平均值,用目的基因的拷贝数除以β-actin 的拷贝数作为靶基因的相对表达量。
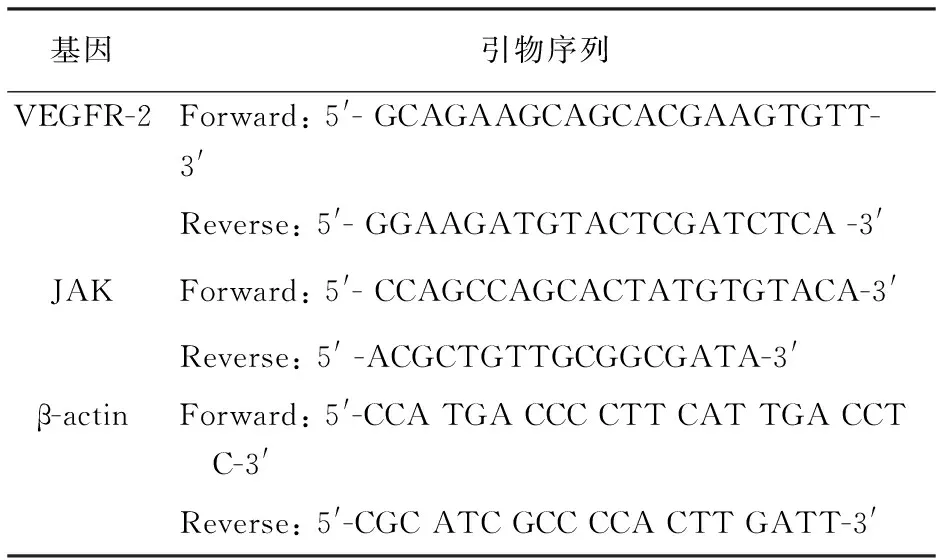
表 1 实时荧光定量PCR扩增基因引物序列
1.3.6 蛋白免疫印迹(Western blot)检测蛋白表达:提取神经元细胞中总蛋白,先进行十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE)电泳。电泳结束后,将凝胶上的蛋白转移到硝酸纤维素膜上。之后将膜放在10 mL封闭液中(2%脱脂奶粉)1 h;加入兔抗鼠p-STAT抗体、含半胱氨酸的天冬氨酸蛋白水解酶3(procaspase 3)和 裂解的含半胱氨酸的天冬氨酸蛋白水解酶3(cleavage caspase 3)(1∶1000),4℃过夜孵育;加入酶标羊抗兔二抗(1∶5000)室温孵育2 h;化学法发光,在暗盒中覆盖X光胶片曝光。胶片拍照,用凝胶图像分析系统分析。
1.4 统计学处理 采用SPSS 13.0统计软件对实验数据进行分析。计量资料采用均值±标准差表示,两组均数间比较采用t检验,多组均数间比较采用单因素方差分析,两两比较采用LSD法。以P<0.05为差异有统计学意义。
2 结果
2.1 低氧预处理促进神经元细胞VEGFR-2表达 与细胞对照组相比较,细胞类缺血组细胞VEGFR-2 mRNA表达下调(P<0.05);与细胞类缺血组相比,低氧预处理组细胞VEGFR-2 mRNA表达上调(P<0.05)(图1)。VEGFR-2蛋白水平的变化与其mRNA水平变化一致(图2)。
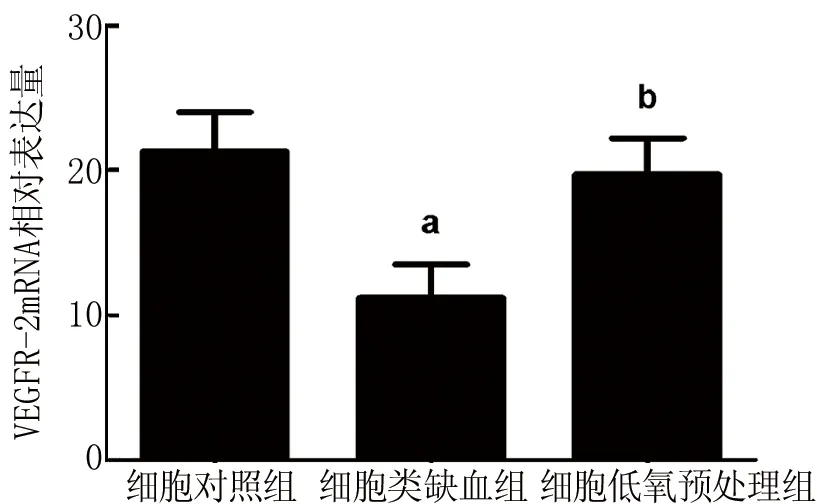
VEGFR-2:血管内皮细胞生长因子受体-2;与细胞对照组比较,aP<0.05;与细胞类缺血组比较,bP<0.05。图2同
图1 qRT-PCR法检测各组小鼠细胞VEGFR-2 mRNA表达

图2 Western blot分析各组小鼠细胞VEGFR-2蛋白表达水平
2.2 低氧预处理促进JAK/STAT信号通路活化 与细胞对照组相比,细胞类缺血组中JAK mRNA和蛋白表达下降,STAT磷酸化受到抑制。与细胞类缺血组相比,细胞低氧预处理组JAK mRNA和蛋白的表达上调(P<0.05),同时也促进了STAT蛋白的活化(P<0.05)(图3、图4)。

与细胞对照组比较,aP<0.05;与细胞类缺血组比较,bP<0.05。图4同
图3 qRT-PCR法检测各组小鼠细胞JAK mRNA表达水平
2.3 低氧预处理对VEGFR-2表达的影响 与转染非特异性siRNA对照组相比,沉默JAK组p-STAT和VEGFR-2表达显著降低(均P<0.05)(图5),表明低氧预处理通过JAK/STAT信号通路调控VEGFR-2的表达。
2.4 低氧预处理促进小鼠海马神经元JAK/STAT信号通路活化和VEGFR-2表达 与动物对照组相比,动物脑缺血组小鼠海马神经元p-STAT和VEGFR-2表达下降(P<0.05);与动物类缺血组相比,低氧预处理则可有效逆转缺氧对p-STAT和VEGFR-2的抑制(P<0.05)(图6)。

图4 Western blot分析各组小鼠细胞JAK和p-STAT蛋白表达水平

VEGFR-2:血管内皮细胞生长因子受体-2,p-STAT:信号转导和转录激活因子蛋白磷酸化,图6同;与非特异转染组比较,aP<0.05
图5 Western blot分析各组小鼠细胞蛋白表达水平
2.5 低氧预处理+VEGF治疗可逆转小鼠神经元细胞凋亡的发生 动物脑缺血+VEGF组小鼠活化的 caspase3表达高于动物对照+VEGF组(P<0.05);而与动物脑缺血+VEGF组相比,动物低氧预处理+VEGF组活化的 caspase3则显著降低(P<0.05)(图7)。
3 讨论
低氧预处理能够诱导产生局灶性缺血耐受,全脑(心)缺血耐受和新生儿缺血耐受[8]。Gidday等首先发现将刚出生的小鼠用含8%(体积分数)O2的低氧条件预处理3 h,在预处理后1 d可显著保护小鼠因缺血损伤诱导的休克[9]。之后,很多实验都对动物低氧预处理进行研究,结果也发现短时间的低氧环境暴露可诱导小鼠产生缺血耐受[10]。研究发现低氧预处理可通过多种分子包括腺苷[11]、钾通道[12]、低氧诱导因子(HIF)[13]、VEGF[14]等诱导耐受。本研究观察到类缺血处理神经元细胞诱导VEGFR-2表达降低,而低氧预处理则促使VEGFR-2表达升高,提示低氧预处理诱导缺血耐受以及对神经元的保护作用与VEGF受体相关。脑缺血耐受的诱导与神经元细胞内多种信号通路的激活及多种蛋白的合成有关。同时研究证据也表明细胞中JAK/STAT信号通路的活化能够影响细胞分裂增殖等重要生理过程[15]。本研究发现类缺血处理降低了神经元中JAK mRNA的表达水平,同时也降低了细胞中p-STAT表达量,与之相反,低氧预处理显著上调JAK表达水平并且也促进p-STAT的表达,表明低氧预处理可活化神经元细胞中JAK/STAT信号通路。

与动物对照组比较,aP<0.05;与动物脑缺血组比较,bP<0.05
图6 Western blot分析各组小鼠海马神经元p-STAT和VEGFR-2蛋白表达水平

与动物对照+VEGF组比较,aP<0.05;与动物脑缺血+VEGF 组比较,bP<0.05
图7 Western blot分析各组小鼠海马神经元procaspase3和cleavage caspase3蛋白表达水平
有研究表明,VEGFR的表达受到JAK/STAT的调控。Dudley等在小血管内皮细胞中发现低氧环境可以诱导JAK/STAT通路的活化并导致细胞VEGFR表达升高[16]。本研究利用siRNA沉默神经元中的靶基因JAK,结果发现细胞经低氧预处理和类缺血处理后,其中p-STAT和VEGFR-2蛋白水平显著降低,表明低氧预处理对VEGFR-2的表达影响可能受JAK/STAT信号通路调控。在小鼠海马神经元中,脑缺血处理降低了细胞的p-STAT和VEGFR-2表达水平,然而低氧预处理显著上调两者的表达量,表明低氧预处理可在小鼠体内诱导JAK/STAT通路活化并诱导VEGFR表达。
某些研究显示注射外源VEGF常用于脑缺血后的神经元保护[17]。Sun等通过在大鼠缺血再灌注1~3 d后脑静脉注射VEGF发现VEGF可显著减少大脑坏死面积,改善神经系统表现,增强新生神经元的存活时间并刺激血管再生[18]。本研究发现低氧预处理+VEGF组中活性caspase 3显著低于脑缺血+VEGF组,其原因可能是低氧预处理上调了神经元细胞中VEGFR-2的表达从而增加了VEGF的治疗效果。
综上所述,本研究结果显示低氧预处理能够通过活化JAK/STAT通路而上调类缺血处理神经元细胞中VEGF受体(VEGFR-2)的表达,这在脑缺血小鼠模型中也得到验证,同时低氧预处理也增强了VEGF对脑缺血小鼠的神经保护作用。低氧预处理诱导缺血耐受的具体机制还不完全清楚,诱导神经保护作用是其中的重要部分,尚需进一步研究证明。
[1]Stevens SL, Leung PY, Vartanian KB, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury [J]. J Neurosci, 2011, 31(23): 8456-8463.
[2]Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning [J]. Cardiovasc Drugs Ther, 2010, 24(3): 225-234.
[3]王震虹, 王祥瑞. 低氧预处理诱导血管内皮生长因子对脑缺血再灌注后损伤及认知功能的影响[J]. 上海医学, 2013, 36(6): 531-535.
[4]An SS, Jin HL, Kim KN, et al. Neuroprotective effect of combined hypoxia-induced VEGF and bone marrow-derived mesenchymal stem cell treatment [J]. Childs Nerv Syst, 2010, 26(3): 323-331.
[5]Zhang HY, Jin XB, Lue TF. Three important components in the regeneration of the cavernous nerve: brain-derived neurotrophic factor, vascular endothelial growth factor and the JAK/STAT signaling pathway [J]. Asian J Androl, 2010, 13(2): 231-235.
[6]Koizumi JYM, Nakazawa T. Experimental studies of ischemic brain edema, a new experimental model of cerebral embolism in rats in which recondition can be introduced in the ischemic area [J]. Jpn J Sturke, 1986, 8(12): 1-8.
[7]Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats [J]. Stroke, 1989, 20 (1): 84-91.
[8]徐恩, 李雯. 脑缺血耐受机制的研究[J]. 中国脑血管病杂志, 2010, 7(2): 86-92.
[9]Gidday JM, Fitzgibbons JC, Shah AR, et al. Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat [J]. Neurosci Lett, 1994, 168(1): 221-224.
[10]李思颉, 张颜波, 邵国, 等. 低氧预适应对小鼠急性脑梗死缺血半暗带血管新生的影响 [J]. 中风与神经疾病杂志, 2012, 29(11): 964-967.
[11]Rytkönen KT, Renshaw GM, Vainio PP, et al. Transcriptional responses to hypoxia are enhanced by recurrent hypoxia (hypoxic preconditioning) in the epaulette shark [J]. Physiol Genomics, 2012, 44(22): 1090-1097.
[12]Wang L, Zhu QL, Wang GZ, et al. The protective roles of mitochondrial ATP-sensitive potassium channels during hypoxia-ischemia-reperfusion in brain [J]. Neurosci Lett, 2011,491(1): 63-67.
[13]Wacker BK, Perfater JL, Gidday JM. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway [J]. J Neurochem, 2012, 123(6): 954-962.
[14]Huang X, Su K, Zhou L, et al. Hypoxia preconditioning of mesenchymal stromal cells enhances PC3 cell lymphatic metastasis accompanied by VEGFR-3/CCR7 activation [J]. J Cell Biochem, 2013, 114(12): 2834-2841.
[15]Lasho T, Tefferi A, Pardanani A. Inhibition of JAK-STAT signaling by TG101348: a novel mechanism for inhibition of KITD816V-dependent growth in mast cell leukemia cells [J]. Leukemia, 2010, 24(7): 1378-1380.
[16]Dudley AX, Thomas D, Best J, et al. A VEGF/JAK2/STAT5 axis may partially mediate endothelial cell tolerance to hypoxia [J]. Biochem J, 2005, 390: 427-436.
[17]Lladó J, Tolosa L, Olmos G. Cellular and molecular mechanisms involved in the neuroprotective effects of VEGF on motoneurons [J]. Front Cell Neurosci, 2013, 7: 181.
[18]Sun Y, Jin K, Xie L, et al. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia [J]. J Clin Invest, 2003, 111(12): 1843-1851.
(本文编辑:时秋宽)
Hypoxia preconditioning activates JAK/STAT signaling and promotes vascular endothelial growth factor receptor-2 expression in neurons
WEIKecong,ZHUYunzhong,LIANGWeibing,ZHANGGaolian*.
*Department of Neurosurgery, the First Affiliated Hospital of Guangxi University of Chinese Medicine, Nanning Guangxi 530023, China
ZHANGGaolian,Email:submission029@gmail.com
ObjectiveToinvestigatetheeffectofhypoxiapreconditioningonJAK/STATsignalingandvascularendothelialgrowthfactorreceptor-2(VEGFR-2)expressionofneurons.MethodsCulturedmouseprimaryneuronsweredividedintocontrol,ischemiaandhypoxiapreconditioninggroup.Neuronsinischemiagroupweretreatedwith95%NO2+5%CO2for30minandcellsinhypoxiapreconditioninggrouppretreatedwith89%N2,l%O2and10%CO2for8times,eachfor20min,followedbyischemiatreatment.Attheendofthetreatment,VEGFR-2mRNAandproteinexpressionsaswellastheexpressionofJAKandphosphorylatedSTAT(p-STAT)inneuronswereanalyzed.Moreover,neuronsweretransfectedwithJAKspecificsiRNApriortohypoxiapreconditioningandischemiatreatment.Then,p-STATandVEGFR-2expressionswereanalyzed.Furthermore,forty-fiveBALB/cmicewererandomizedintocontrol(n=8),control+VEGF(n=7),ischemia(n=8),ischemia+VEGF(n=7),hypoxiapreconditioning(n=8),hypoxiapreconditioning+VEGFgroups(n=7).Afterthetreatment,neuronsinhippocampuswereseparatedfortheanalysisofp-STATandVEGFR-2expressionsalongwiththeactivationofcaspase3protein.ResultsComparedwiththecontrol,theexpressionsofVEGFR-2mRNAweremarkedlydownregulated(P<0.05)byischemia.Comparedwiththeischemicgroup,hypoxiapreconditioningsignificantlyelevatedtheexpressionsofVEGFR-2mRNA,JAKmRNAandprotein,andthephosphorylationofSTAT.ComparedwithnonspecificsiRNAgroup,knockdownofJAKinhibitedthelevelsofp-STATandVEGFR-2expression.Inmousemodels,ischemiasignificantlydownregulatedtheexpressionsofp-STATandVEGFR-2,whichwerereversedbyhypoxiapreconditioning.Comparedwithischemia+VEGFgroup,hypoxiapreconditioning+VEGFremarkablyinhibitedtheactivationofcaspase3.ConclusionsHypoxiapreconditioningmightactivateJAK/STATandinduceVEGFR-2expressions,leadingtoenhancementofneuroprotectioninducedbyVEGFinneuronsafterischemiatreatment.
brain,hypoxia;ischemicpreconditioning;intracellularsignalingpeptidesandproteins;januskinases;JAK/STAT;vascularendothelialgrowthfactorreceptor-2
10.3969/j.issn.1006-2963.2015.01.010
530023 广西中医药大学第一附属医院神经外科
张高炼,Email: submission029@gmail.com
R743.3
A
1006-2963 (2015)01-0040-06
2014-05-30)
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2022年8期)2022-12-07
温州大学学报(自然科学版)(2022年2期)2022-05-30
昆明医科大学学报(2021年8期)2021-08-13
天津医科大学学报(2021年3期)2021-07-21
潍坊学院学报(2020年2期)2021-01-18
昆明医科大学学报(2020年11期)2020-12-28
中国特种设备安全(2019年4期)2019-05-20
浙江医学(2018年16期)2018-09-08
制导与引信(2017年3期)2017-11-02
军事体育学报(2015年2期)2015-02-27
