
超高效及高效液相色谱法测定华法林钠片含量的结果比对
2015-06-07 09:15丁大中宋梓瑜
中国药业 2015年14期
丁大中,宋梓瑜,于 毓
(山东省青岛市食品药品检验检测中心,山东 青岛 266071)
华法林钠为双香豆素的衍生物,临床应用广泛,其优点是作用强且稳定、可口服给药、应用方便、价廉且作用持久[1-2]。目前,进口华法林钠片采用《国家食品药品监督管理局进口药品注册标准》[3]检验,含量测定采用高效液相色谱(HPLC)法。超高效液相色谱(UPLC)法比HPLC法具有高效、快速、灵敏度和分辨率高、微量、经济及环境友好等优点[4-8],但将其用于华法林钠含量测定还未见报道。本研究参考文献[9-12],成功建立了测定华法林钠片含量的UPLC法,该方法操作灵敏、准确,且能实现高通量的分析样品。
1 仪器与试药
Agilent 1200型高效液相色谱仪(美国Agilent公司);Waters Acquity UPLC超高效液相色谱仪(包含Empower色谱工作站,美国Waters公司);AG285型十万分之一天平(瑞士 Mettler Toledo公司);超纯水仪(美国Millipore公司)。华法林钠对照品(中国食品药品检定研究院,批号为101163-201001,含量为92.3%);磷酸(天津市科密欧化学试剂有限公司,批号为20120104)、甲醇(德国Merch公司)、乙腈(德国Merch公司)均为色谱纯;超纯水(Millipore超纯水仪公司);华法林钠片(芬兰Orion Corparation公司,规格为每片 3 mg,批号分别为 1555638,1555639,1556716,1556720,1556722)。
2 方法与结果
2.1 色谱条件
HPLC色谱条件:色谱柱为Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5μm);以水(磷酸调 pH 至 3.0)为流动相 A、乙腈为流动相B,进行梯度洗脱;流速1.5 mL/min;柱温30℃;检测波长280 nm;进样量 20 μL。
UPLC色谱条件:色谱柱为Thermo Syncronis C18柱(100 mm×2.1 mm,1.7μm);以水(磷酸调 pH 至 3.0)为流动相 A、乙腈为流动相B,进行梯度洗脱;流速0.5 mL/min;柱温30℃;检测波长280 nm;进样量5 μL。梯度洗脱方法见表1。
2.2 溶液制备
精密称取华法林钠对照品10 mg,置100 mL容量瓶中,加溶剂A(水∶乙腈=2∶8)振摇溶解并稀释至刻度,摇匀,即得对照品溶液(100 μg/mL)。取华法林钠片20片,精密称定总质量,研细,精密称取细粉(约相当于华法林钠5 mg),置50 mL容量瓶中,加溶剂A适量,超声10 min使溶解并稀释至刻度,摇匀,离心,取上清液作为供试品溶液(100 μg/mL)。

表1 HPLC法与UPLC法梯度洗脱方法
2.3 UPLC方法学考察
线性关系考察:精密吸取 2.2 项下对照品溶液 3,4,5,6,8 μL,注入超高效液相色谱仪,按UPLC色谱条件进样测定。以样品峰面积(Y)为纵坐标、进样量(X,μg)为横坐标进行线性回归,得回归方程 Y=4.36×106X -1.53×104,r=0.999 8(n=5)。结果,华法林钠进样量在0.284~0.758 μg范围内与峰面积线性关系良好。
最低检测限确定:以信噪比为3∶1(S/N=3)的浓度进样,华法林钠的最低检测限为0.02 ng。
精密度试验:取2.2项下同一对照品溶液,连续进样6次。结果华法林钠保留时间的 RSD为 0.06%,峰面积的 RSD为0.08%(n=6),表明仪器精密度良好。
重复性试验:精密称取同一批(批号为1555638)样品粉末6份,制备供试品溶液,按UPLC色谱条件进样测定。结果的 RSD为0.28%(n=6),表明方法重复性良好。
稳定性试验:取2.2项下制备的同一供试品溶液,分别于0,2,4,8,16,24 h按拟定色谱条件依法进样 6次。结果峰面积的RSD为0.21%(n=6),表明供试品溶液在24 h内稳定。
加样回收试验:精密称取半量已知含量的同一批(批号为1555638)样品细粉9份,按2.2项下方法制备供试品溶液,分别精密加入0.51 g/L华法林钠对照品溶液,稀释至刻度,摇匀,依法进样测定,计算回收率。结果见表2。
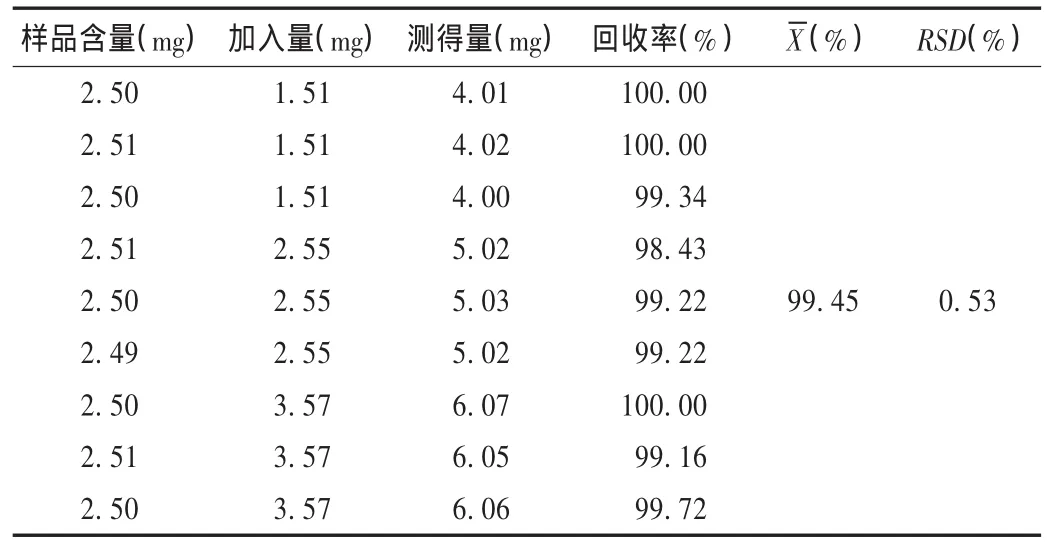
表2 华法林钠加样回收试验结果(n=9)
系统适用性试验:精密称取4-羟基香豆素和(E)-4-苯基-3-丁烯-2-酮对照品各10 mg,置100 mL容量瓶中,加溶剂A溶解,精密量取1 mL,置100 mL容量瓶中,精密加入2.2项下供试品溶液5 mL,加溶剂A溶解,精密量取5 mL置500 mL容量瓶中,加溶剂A稀释至刻度,即得供试品溶液。各组分的洗脱顺序为4-羟基香豆素、(E)-4-苯基-3-丁烯-2-酮和华法林钠,前两者峰的分离度应不小于2.5。结果及色谱图见表3和图1。

表3 HPLC法与UPLC法系统适用性试验结果对比
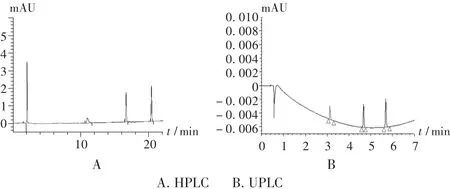
图1 系统适用性试验色谱图
2.4 样品含量测定
取5批样品,按2.2项下方法分别制备对照品和供试品溶液,分别按2.1项下HPLC和UPLC色谱条件进样测定,用外标法以峰面积计算含量。结果表明,在UPLC色谱条件下,华法林钠峰形良好,与杂质峰的分离度高,且UPLC和HPLC方法所得结果一致,见表4。HPLC法与UPLC法中华法林钠对照品与供试品的典型色谱图见图2。
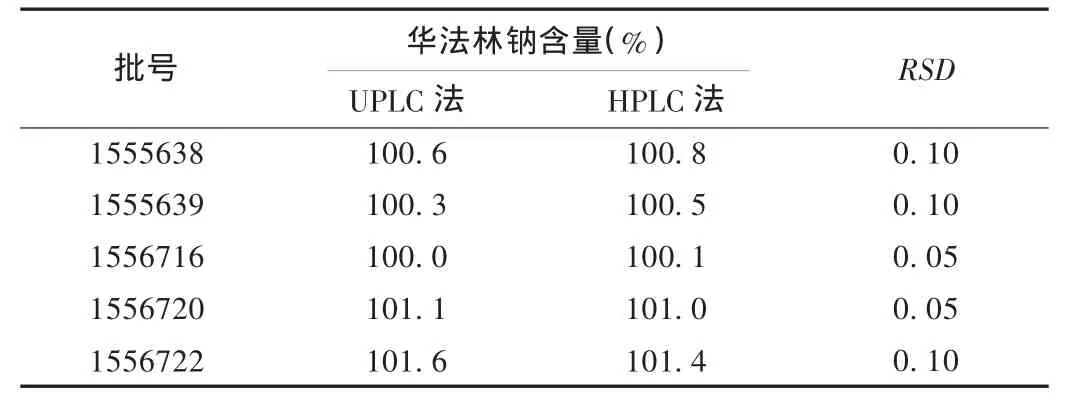
表4 UPLC法和HPLC法含量测定结果对比(n=5)
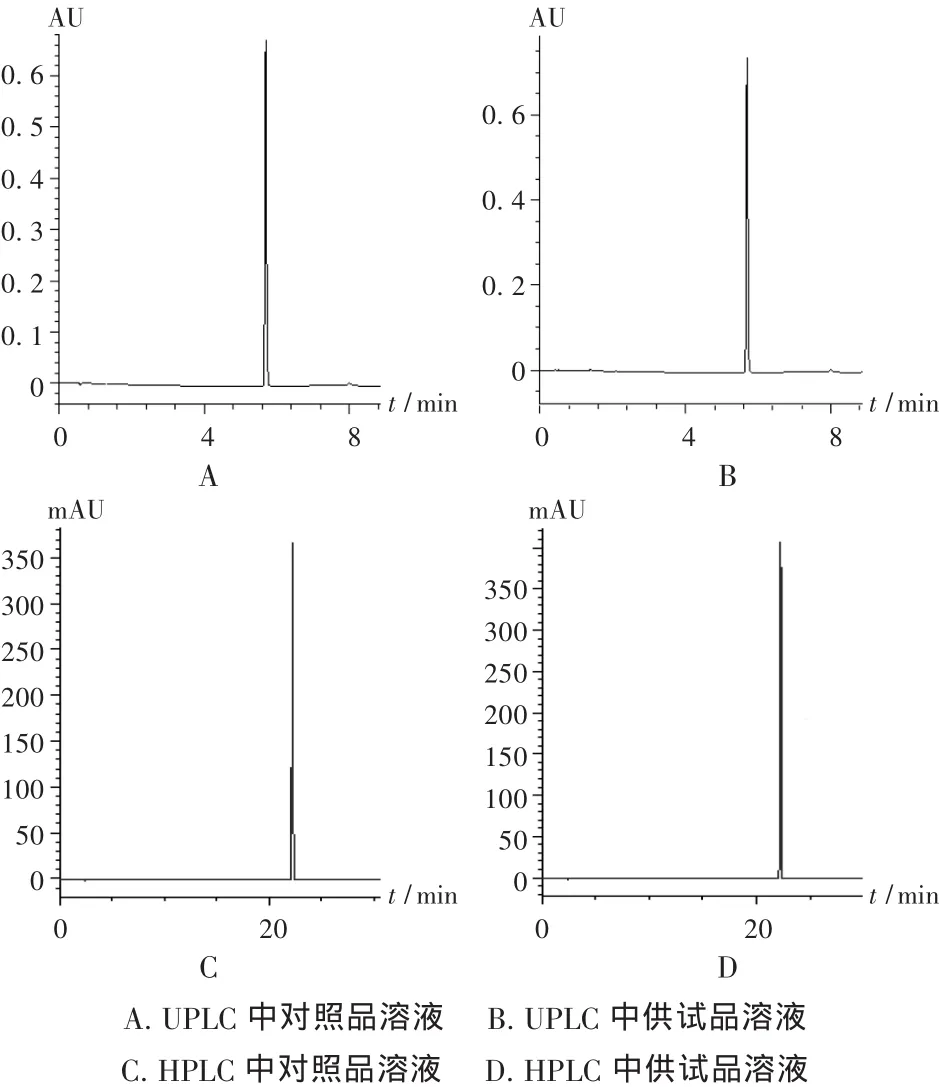
图2 典型色谱图
3 讨论
梯度洗脱方法优化:原标准中HPLC法梯度洗脱时间为50min,流速为1.5 mL/min,时间长,流速大,消耗大量有机溶剂。采用UPLC 法将洗脱时间缩短至 12.5 min,流速降至 0.5 mL/min,时间和流速均减少了70%以上,实现快速测定含量,同时减少溶剂消耗量,极大地降低了成本,减少了环境污染。
流速选择:本研究中尝试了多种流速后发现,流速小时,华法林钠出峰时间长,使梯度洗脱时间过长,达不到快速分离的目的;流速大时,系统和色谱柱反压过大,无法正常分析样品;最后确定流速为0.5 mL/min,在系统可承受压力范围内和在保证分离效果的前提下,尽可能地缩短了梯度洗脱时间。
进样量选择:试验确定了方法的线性范围,并对系统精密度、重复性、稳定性和回收率进行了考察,结果显示,进样量小会降低方法的精密度和重复性,进样量大会超出线性范围,出现过载现象。最终确定了进样为5 μL。
系统适用性试验:由结果可见,2种方法均能良好地达到标准规定的要求,但UPLC法峰形更好,各组分间的分离度更优。
本研究中建立的UPLC法相比HPLC法用于华法林钠片的含量测定,具有检测效率高、灵敏度好、准确快速且绿色环保的优点,并可推广用于华法林钠其他制剂的含量测定。
[1]张石革,马国辉,臧 靖.抗血栓药华法林钠的进展与合理应用[J].中国全科医学,2005,8(20):1 680.
[2]郑必龙,刘 俊.华法林抗凝血作用及影响因素分析[J].安徽医药,2013,17(11):1 975 -1 977.
[3]JX20120121,国家食品药品监督管理局进口药品注册标准[S].
[4]Wirth MJ.Mass transport in sub-2-μm high-performance liquid chromatography[J].J Chromatogr A,2007,1 148(1):128.
[5]杨义芳.超高效/高分离度快速/超快速液相色谱在中药及其制剂研究中的应用[J].中草药,2008(8):1 259.
[6]陈 佳,王钢力,姚令文,等.超高效液相色谱(UPLC)在药物分析领域中的应用[J].药物分析杂志,2008,28(11):1 976 -1 981.
[7]张承军,王秀梅,王建良,等.UPLC法测定生长抑素原料药的含量[J].西部中医药,2012,25(10):37-40.
[8]Gumustas M,Kurbanoglu S,Uslu B,et al.UPLC versus HPLC on drug analysis:advantageous,applications and their validation paramaters[J].Chromatographia,2013,76(21 -22):1 365 -1 427.
[9]王 慧,况 刚,封海霞,等.高效和超高效液相色谱法测定乐脉丸中丹酚酸B含量的比较[J].中国药业,2012,21(22):53-54.
[10]谢新民,刘志刚,黄雪峰,等.高效液相色谱法测定盐酸普萘洛尔乳膏的含量[J].中国药业,2015,24(3):23 -24.
[11]李 霞,李 继,戴 涌,等.高效液相色谱法测定复方甘草麻黄碱片中盐酸麻黄碱含量[J].中国药业,2015,24(1):43-44.
[12]逯小萌.高效液相色谱法测定氨糖美辛肠溶片中吲哚美辛含量[J].中国药业,2014,23(24):74 -75
猜你喜欢
分析仪器(2022年5期)2022-10-14
中国石油大学学报(自然科学版)(2022年4期)2022-09-05
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
化工管理(2021年7期)2021-05-13
中国应急管理科学(2021年4期)2021-04-13
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
