
铁原子与NO反应的密度泛函理论研究
2015-03-22 10:43戚越舟苏亚欣
原子与分子物理学报 2015年6期
戚越舟, 苏亚欣
(东华大学环境科学与工程学院, 上海 201620)
铁原子与NO反应的密度泛函理论研究
戚越舟, 苏亚欣
(东华大学环境科学与工程学院, 上海 201620)
采用密度泛函理论(DFT)计算研究铁原子与NO反应的相关微观反应机理.全参数优化了四重态和六重态反应势能面上各驻点的几何结构,用频率分析方法和内禀反应坐标(IRC)方法对过渡态进行了验证,得到了该反应的反应势能面曲线,并讨论了势能面的交叉情况.结果表明,该反应为典型的两态反应,反应通道一中出现了一个势能交叉点,反应通道二中出现了两个势能交叉点,反应通道三中出现了三个势能交叉点.势能面上的交叉点能够有效降低反应的活化能,增加反应放热,这在动力学和热力学上都是有利的.
过渡金属原子; 量子化学; 反应微观机理; 势能交叉点
1 引 言
近20年来,探索金属催化剂的内部电子特性,动力特性已经成为一个非常活跃的领域.而过渡金属由于多相催化的特性而倍受关注,由此展开了大量的实验和理论研究[1,2].例如,对于当前煤燃烧过程中排放NOX的环境污染问题,大量的研究表明金属能有效促进催化还原NO,包括K、Na、Ca等主族金属以及Cu、Co、Ni、Fe等过渡金属[3-7].在众多的过渡金属中,铁系催化剂是一种有效脱除氮氧化物的理想催化剂,前期的大量的实验研究[8-10]表明金属铁直接催化还原NO是一种高效的脱硝方法.Blagojevic等[11]研究了Fe+催化CO还原N2O的反应路径,Francesca Rondinelli等[12]通过DFT理论也得到了Fe+和Mn+催化CO脱除N2O的反应路径,均发现Fe+能有效降低反应的活化能.西北师范大学王永成等[13,14]研究了Fe+,FeO+消除N2O,发现铁系催化剂对于反应的决速态起关键的作用.但是当前绝大多数关于铁系催化剂脱除氮氧化物的量化模拟都集中在铁离子,铁的氧化物离子上,对于铁原子本身的研究,Andreas Fiedler等[15]研究了Fe、N、O三种元素组成的同分异构体,但是缺少了反应的路径和动力学的研究.本文以Fe+NO为研究体系,用密度泛函理论(DFT)计算方法,研究了反应体系在四、六重态势能面上的反应机理,该研究对于人们理解金属铁催化脱硝的重要反应提供了理论依据.
2 计算方法
密度泛函理论(DFT)已广泛用于过渡金属化学的理论计算,计算结果的可靠性已被研究学者所公认[16].文中采用Becke三参数交换泛函,并结合LYP相关泛函(即B3LYP方法)[17],对Fe,N,O采用6-31G(d)基组,对反应体系势能面上的所有反应物、中间体、过渡态和产物的几何构型进行了全参数优化,通过频率分析证实了各反应物、中间体和产物的能量为局部极小,各过渡态有唯一振动虚频.对各势能面上的每一个鞍点进行了内禀反应坐标(IRC)计算,确认了每个基元步骤过渡态的可靠性.为了获取更为精确的相对能量值,在此几何构型基础上进一步采用B3LYP/6-311+G(d,p)方法进行单点能计算.
本文所选用的计算方法广泛用于体系中含有过渡金属的电子结构计算,Qiao Sun等[18]通过此计算方法研究铁簇催化甲烷,Lichen Wang等[19]通过该方法研究了Fe(NO)n+的特性,Q. Sun等[20]通过该方法研究了纳米铁簇的特性,大量的模拟计算证明这是一种计算耗时合理,计算准确的方法[21,22].本文所有计算都在Gaussian 09程序中完成.
3 结果与讨论
本文着重对脱硝基元反应(Fe+NO→FeO+1/2N2)进行深入的研究.有过渡金属参与的反应中,高自旋态过渡金属复合物常常具有多个未成对电子,由于受配体与金属d轨道之间电子的交换作用等因素的影响,导致过渡金属在催化反应过程中很可能发生自旋翻转而出现势能面交叉现象,即在不同自旋多重度的两个势能面的交叉区附近出现自旋翻转[23-25].本文以铁原子与NO的反应为研究体系,用密度泛函理论(DFT)计算方法,研究了反应体系在四、六重态势能面上的反应机理,分别优化了四重态和六重态反应势能面上所有驻点的几何构型,得到基元反应的微观进程,结果如图1~6所示,四重态的构型命名为C,TS,六重态的构型命名为C`,TS`.其中所有的中间体以及过渡态均为平面结构,键角的单位为度,键长的单位为埃.表1为各反应过渡态及中间体振动频率分析的结果.表2为反应通道上各驻点的能量,其中,Eb3lyp表示采用6-311+G(d,p)方法计算得到的节点能;Ezpe为零点能;Etotal为各驻点的总能量;Erel为相对能量.
表1 各反应的中间体和过渡态的振动频率
Table 1 vibration frequency of the intermediates and transition states for each reaction channel
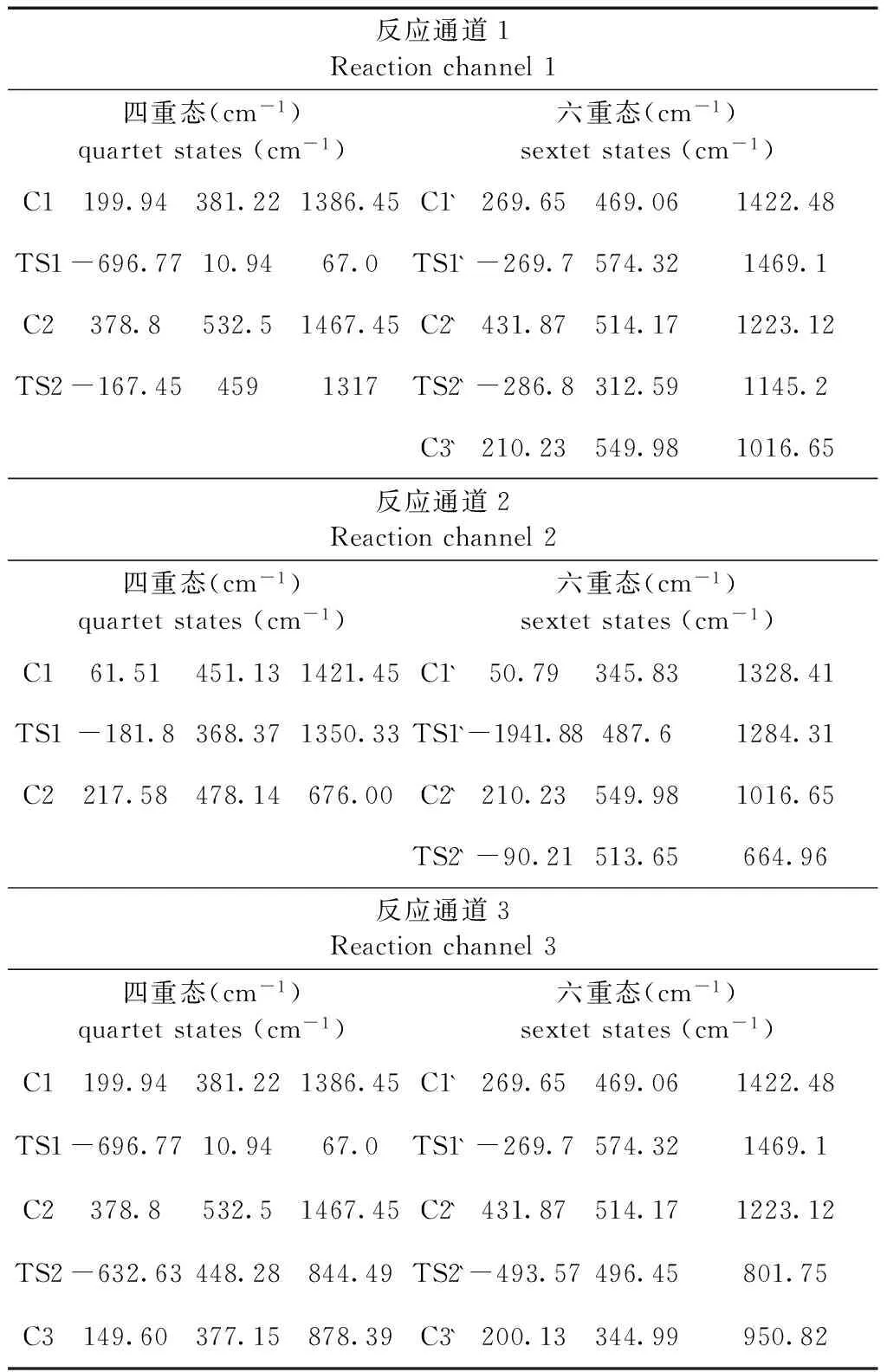
反应通道1Reactionchannel1四重态(cm-1)quartetstates(cm-1)六重态(cm-1)sextetstates(cm-1)C1199.94381.221386.45C1`269.65469.061422.48TS1-696.7710.9467.0TS1`-269.7574.321469.1C2378.8532.51467.45C2`431.87514.171223.12TS2-167.454591317TS2`-286.8312.591145.2C3`210.23549.981016.65反应通道2Reactionchannel2四重态(cm-1)quartetstates(cm-1)六重态(cm-1)sextetstates(cm-1)C161.51451.131421.45C1`50.79345.831328.41TS1-181.8368.371350.33TS1`-1941.88487.61284.31C2217.58478.14676.00C2`210.23549.981016.65TS2`-90.21513.65664.96反应通道3Reactionchannel3四重态(cm-1)quartetstates(cm-1)六重态(cm-1)sextetstates(cm-1)C1199.94381.221386.45C1`269.65469.061422.48TS1-696.7710.9467.0TS1`-269.7574.321469.1C2378.8532.51467.45C2`431.87514.171223.12TS2-632.63448.28844.49TS2`-493.57496.45801.75C3149.60377.15878.39C3`200.13344.99950.82
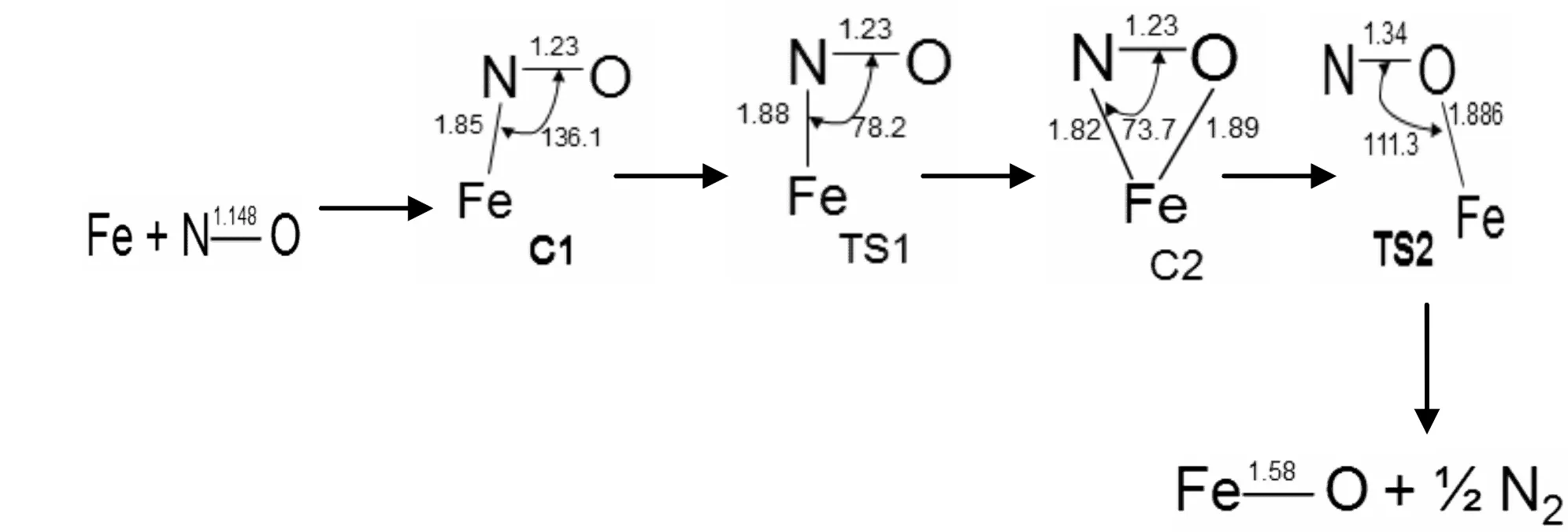
图1 反应通道一四重态反应势能面上所有驻点的构型及反应的微观进程Fig. 1 Optimized geometrical configurations of various species and micro-reaction pathways in the reaction of channel 1 for quartet state

图2 反应通道一六重态反应势能面上所有驻点的构型及反应的微观进程Fig. 2 Optimized geometrical configurations of various species and micro-reaction pathways in the reaction of channel 1 for sextet state
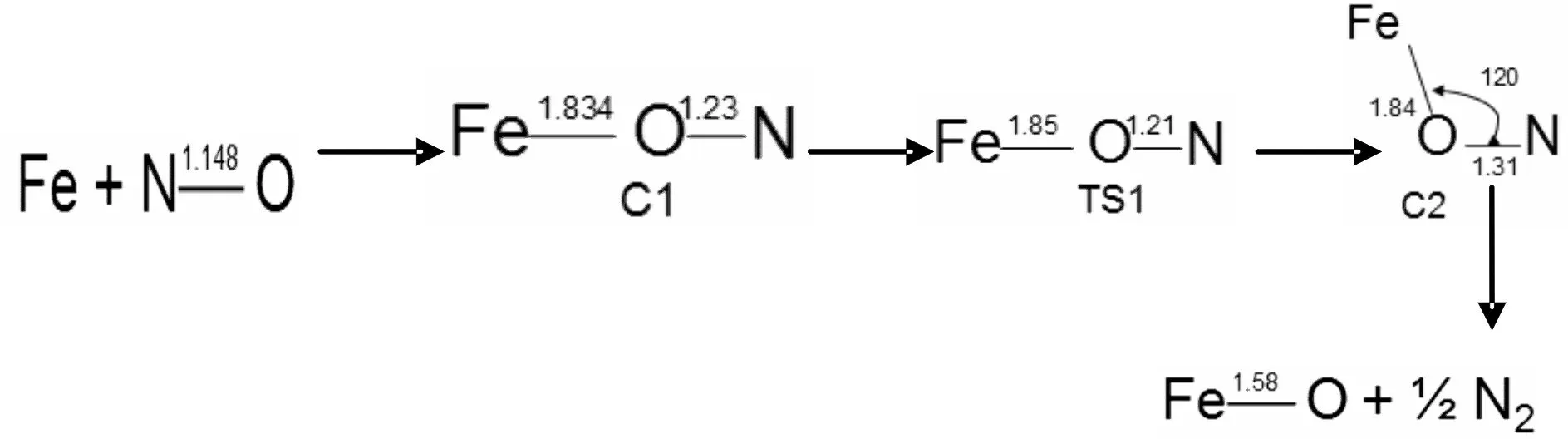
图3 反应通道二四重态反应势能面上所有驻点的构型及反应的微观进程Fig. 3 Optimized geometrical configurations of various species and micro-reaction pathways in the reaction of channel 2 for quartet state
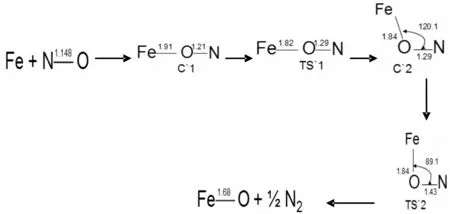
图4 反应通道二六重态反应势能面上所有驻点的构型及反应的微观进程Fig. 4 Optimized geometrical configurations of various species and micro-reaction pathways in the reaction of channel 2 for sextet state
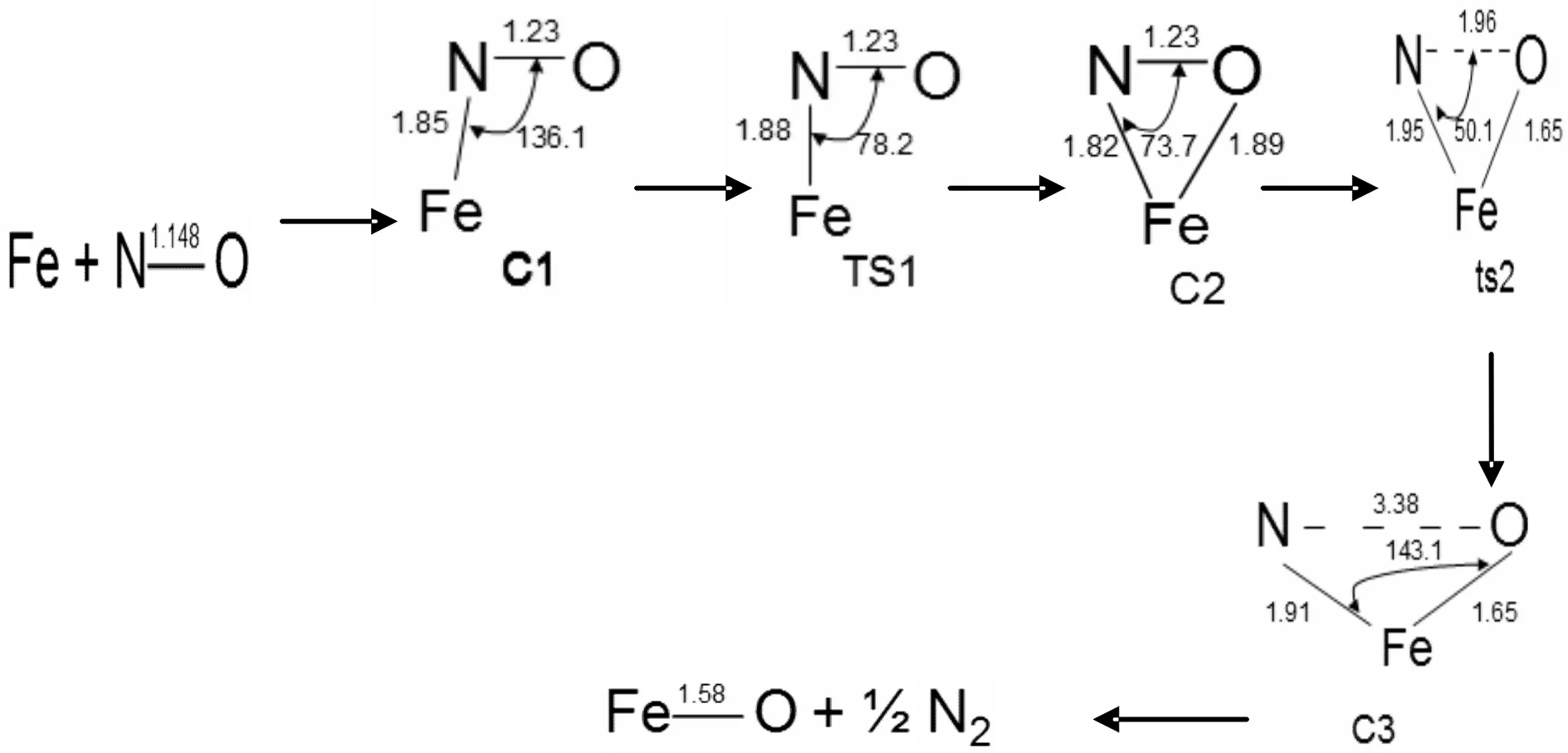
图5 反应通道三四重态反应势能面上所有驻点的构型及反应的微观进程Fig. 5 Optimized geometrical configurations of various species and micro-reaction pathways in the reaction of channel 3 for quartet state
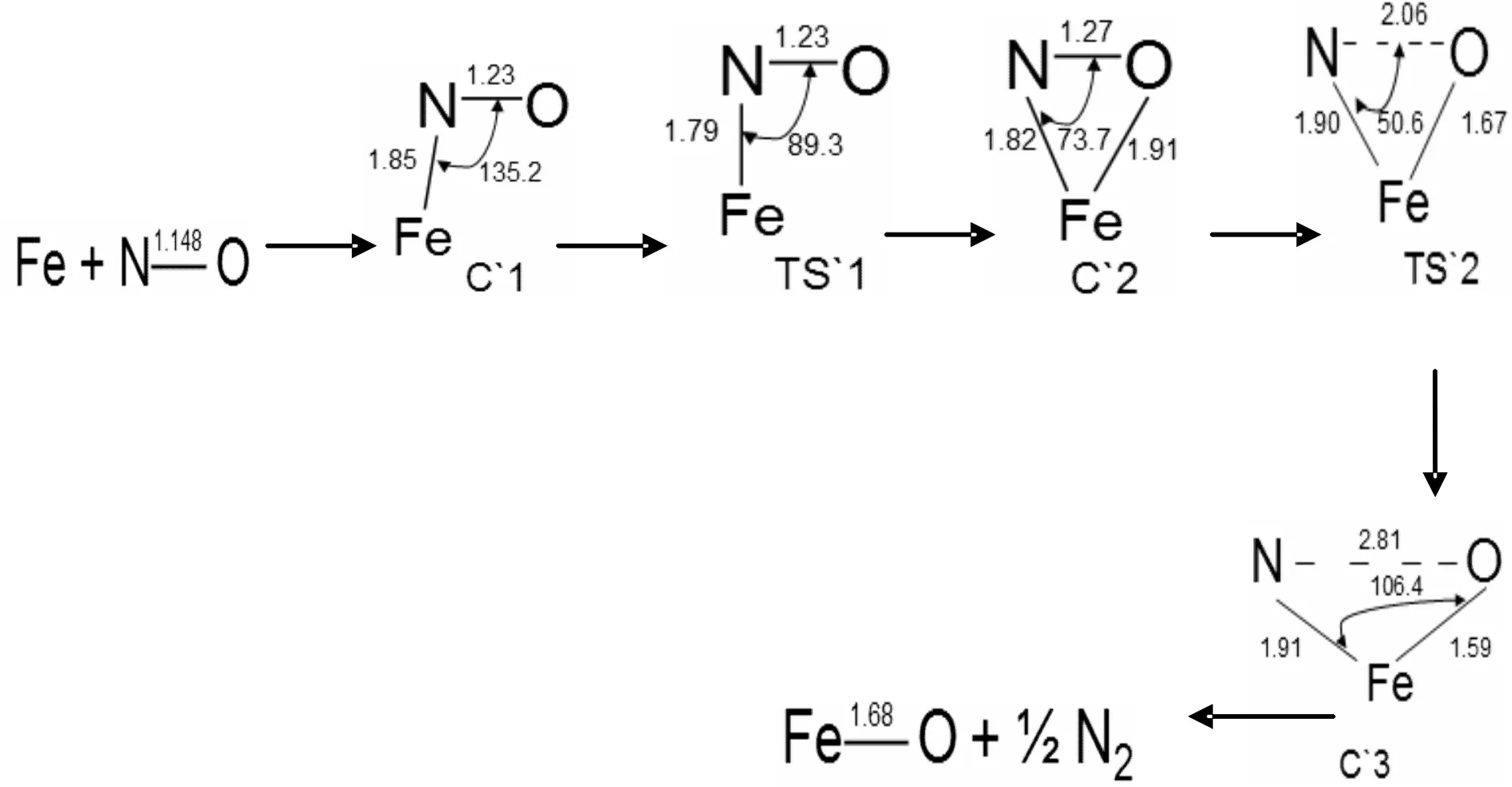
图6 反应通道三六重态反应势能面上所有驻点的构型及反应的微观进程Fig. 6 Optimized geometrical configurations of various species and micro-reaction pathways in the reaction of channel 3 for sextet state
3.1 反应通道一
图1、2所示为反应通道一中Fe原子与NO在四,六重态反应势能面上所有驻点的构型及反应的微观进程.首先,铁原子进攻NO的N端生成相应的反应初始复合物C1和C1`,此过程无需翻越任何势垒.在四重态反应势能面上形成Fe,N,O键角呈136.1°的中间体C1,属于Cs点群,电子组态为4A″,Fe-N键键长为1.85埃,N-O键键长由1.14埃伸长到1.23埃,键级降低.这表明随着Fe-N化学键的形成,N-O键逐渐变弱,键级的减小有利于N-O键的断裂.六重态反应势能面上也发生类似的反应,形成Fe,N,O键角呈135.2°的中间体C1`,属于Cs点群,电子组态为6A″,Fe-N键键长为1.85埃,N-O键键长由1.14埃伸长到1.23埃.接着C1,C1`沿着反应路径,经过相应的过渡态TS1和TS1`,生成三角形状产物复合物C2和C2`.在这个过程中,四重态反应势能面上C1需要克服8.97 Kcal/mol的势垒,Fe,N,O键角由136.1°减小到73.7°,同时生成了键长为1.89埃的Fe-O键.六重态反应势能面上的过程也相似,但是C1`需要克服38.1 Kcal/mol的势垒,生成的Fe-O键键长为1.91埃.由于四重态反应势能面上的势垒低,所以反应更加容易反应.最后,C2沿着反应路线,经过过渡态TS2,N-O键最终断开生成FeO和1/2的N2.而六重态势能面上的C2`则需再经历一个过渡态TS2`生成C3`,N-O键键长由1.27埃伸长至1.31埃,键级降低,Fe-O键由1.91埃缩短至1.85埃,同时Fe,O,N的键角增大到120°,此过程需要克服17.5Kcal/mol的势垒.C`3最终分解成FeO和1/2的N2.从图7中可以看出,反应初始阶段六重态势能面上的中间体C1`比四重态上的中间体C1稳定,C1的相对能量比C1`高4.43 Kcal/mol.初始阶段,反应更偏向高自旋态.之后六重态的势能面总是高于四重态,六重态的能量比四重态的高,反应主要是通过四重态的反应路径发生.
3.2 反应通道二
图3、4所示为反应通道二中铁原子与NO在四,六重态反应势能面上所有驻点的构型及反应的微观进程.首先,铁原子进攻NO的O端生成反应初始复合物C1和C1`,此过程无需翻越任何势垒.在四重态反应势能面上形成Fe,N,O键角呈179.5°的中间体C1,属于Cs点群,电子组态为4A″,Fe-O键键长为1.83埃,O-N键键长由1.14埃伸长到1.23埃,键级降低.这表明随着Fe-O化学键的形成,O-N键逐渐变弱,键级的减小有利于O-N键的断裂.六重态反应势能面上也发生类似的反应,形成Fe,N,O键角呈179.5°的中间体C1`,属于Cs点群,电子组态为6A″,Fe-O键键长为1.91埃,N-O键键长由1.14埃伸长到1.21埃.接着C1,C1`沿着反应路径,经过相应的过渡态TS1和TS1`,生成复合物C2和C2`.在四重态反应势能面上,C1需要克服22.1 Kcal/mol的势垒,Fe,N,O键角由179.5°减小到120.1°,Fe-O键键长由1.23埃伸长至1.31埃,这个过程需要克服22.1 Kcal/mol的势垒,最终中间体C2的O-N键断裂生成了FeO和1/2的N2;在六重态反应势能面上也发生类似的反应,而C1`需要克服6Kcal/mol的势垒,相较而言六重态反应势能面上此过程更加容易发生.最后C2`沿着反应路径,经过过渡态TS2`,O-N断裂,最终生成了FeO和1/2的N2.从图8中可以看出,反应初始阶段四重态势能面上的中间体C1比六重态上的中间体C1`稳定,C1的相对能量比C1`高13.3 Kcal/mol.初始阶段,反应更偏向低自旋态.之后四重态的势能面总是高于六重态,四重态的能量比六重态的高,反应主要是通过六重态的反应路径发生.
3.3 反应通道三
图5、6所示为反应通道三中铁原子与NO在四,六重态反应势能面上所有驻点的构型及反应的微观进程.三角形状产物之前的反应大致与反应通道一相同.之后C2,C2`沿着反应路径,经过相应的过渡态TS2和TS2`,N-O键断裂,在四重态反应势能面上N,O的距离为3.38埃,Fe-O键键长由1.89埃缩短至1.65埃,Fe-N键键长由1.82埃伸长至1.91埃,键级降低.Fe,O,N之间的键角扩大至143.1°.这个过程使N-O键彻底断裂,需要克服47.74 Kcal/mol的势垒.最终中间体C3 的Fe-N键断裂生成了FeO和1/2的N2.六重态反应势能面上也发生类似的反应.复合物C3`,N,O之间的距离由1.27埃伸长至2.81埃,Fe-O键键长由1.91埃缩短至1.59埃,Fe-N键键长由1.82埃伸长至1.91埃,键级降低.Fe,O,N之间的键角扩大至106.4°,需要克服68.7 Kcal/mol的势垒,相较而言此过程四重态反应势能面上更加容易发生.我们发现C3和C3`的构型变化很大原因很有可能是由于亲电子性和极化的作用.从图8中可以看出,反应通道三的反应比较复杂,势能面上有多个交叉点,分别是在不同势能面上反应.
对反应路径上的所有驻点进行了振动频率分析,结果如表1所示,研究的振动分析计算结果表明:各反应物、产物和中间体的振动分析结果是力常数矩阵本征值全为正,说明它们为势能面上的稳定点.各过渡态的振动分析结果,力常数矩阵本征值均有且仅有唯一的负值.同时采用IRC计算验证了过渡态的可信性,结果表明优化得到的中间体和过渡态都是合理且可信的.
3.4 反应势能面的交叉点分析
为了更清楚了解Fe与NO的反应机理,我们进一步探讨反应在四重态和六重态势能面上的交叉行为如图7~图9所示.反应通道一中:六重态中间体C1`比四重态中间体C1的能量低4.43 Kcal/mol,六重态过渡态TS1`比四重态过渡态TS1的能量高24.7 Kcal/mol;反应通道二中:四重态中间体C1比六重态中间体C1`的能量低13.3 Kcal/mol,四重态过渡态TS1比六重态过渡态TS1`的能量高2.1 Kcal/mol,四重态产物能量比六重态产物的能量高25.1 Kcal/mol;反应通道三中:六重态中间体C1`比四重态中间体C1的能量低4.43 Kcal/mol,六重态过渡态TS1`比四重态过渡态TS1的能量高20.2 Kcal/mol,六重态中间体C2`比四重态中间体C2的能量低2.45 Kcal/mol,六重态过渡态TS2`比四重态过渡态TS2的能量18.5 Kcal/mol,六重态中间体C3`比四重态中间体C3的能量高8.9 Kcal/mol.这就大概确定了反应可能在反应通道一C1`→TS1,反应通道二C1→TS1`,C2→反应产物,反应通道三C1`→TS1,C2→TS2`,TS2`→C3的过程中发生了“系间窜越”,使得反应在不同势能面间发生了翻转.反应通道一中起初在六重态势能面上进行,然后经过翻转到四重态势能面进行;反应通道二起初在四重态势能面上进行,然后经过翻转到六重态势能面进行,最终生成四重态反应产物;反应通道三中起初在六重态势能面上进行,然后经过一次翻转到四重态势能面进行,接着第二次翻转到在六重态势能面上,最终生成四重态产物.根据Hammond假设,这是一个典型的“两态反应”[25].反应通道一中的势能交叉点有效的降低活化能24.7 Kcal/mol,同时增加反应放热29.5 Kcal/mol. 反应通道二中的第一个势能交叉点有效降低活化能2.8 Kcal/mol,第二个势能交叉点增加反应放热25.1 Kcal/mol.反应通道三中第一个势能交叉点有效降低活化能20.2 Kcal/mol;第二个势能交叉点有效降低活化能18.5 Kcal/mol;第三个势能交叉虽然不能有效降低反应活化能但增加反应放热25.1 Kcal/mol.这显然在动力学和热力学上都是有利的.
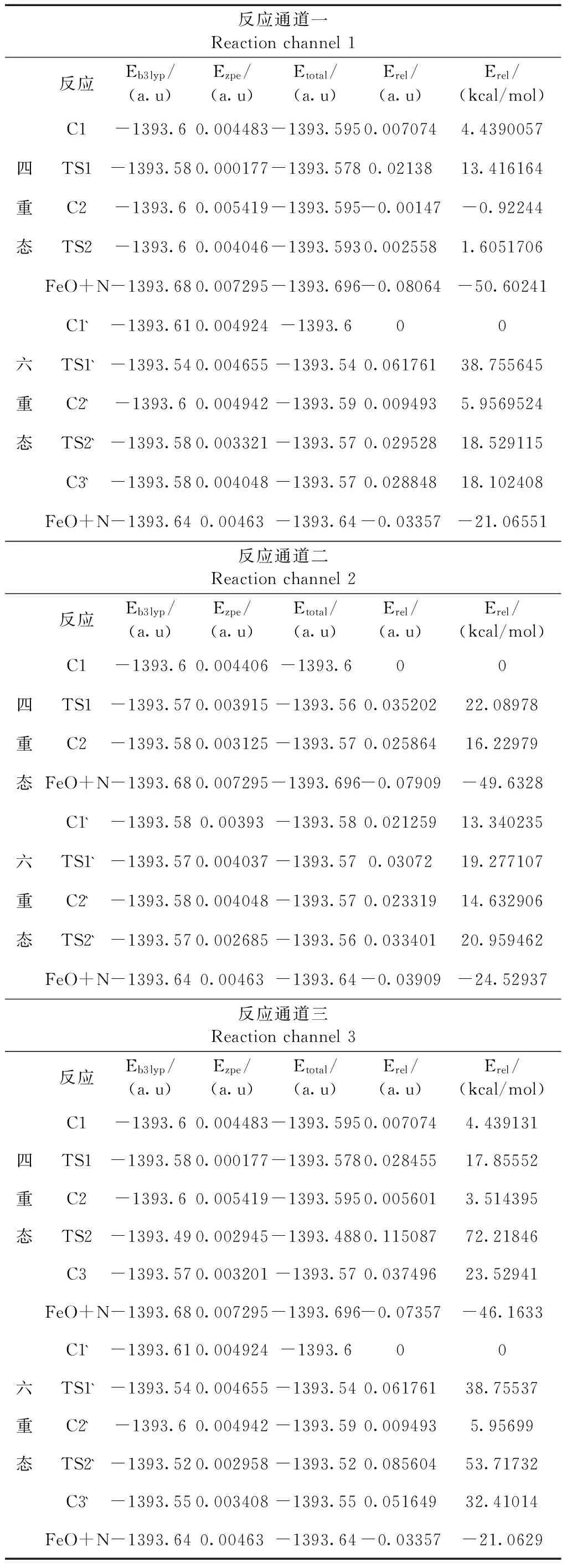
表2 反应通道上各驻点的能量
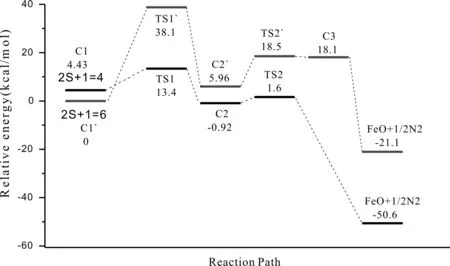
图7 Fe+NO在反应通道一中四重态和六重态的反应势能面图Fig. 7 Diagram of PESs for the reaction of Fe+NO on the quartet and sextet states in channel 1
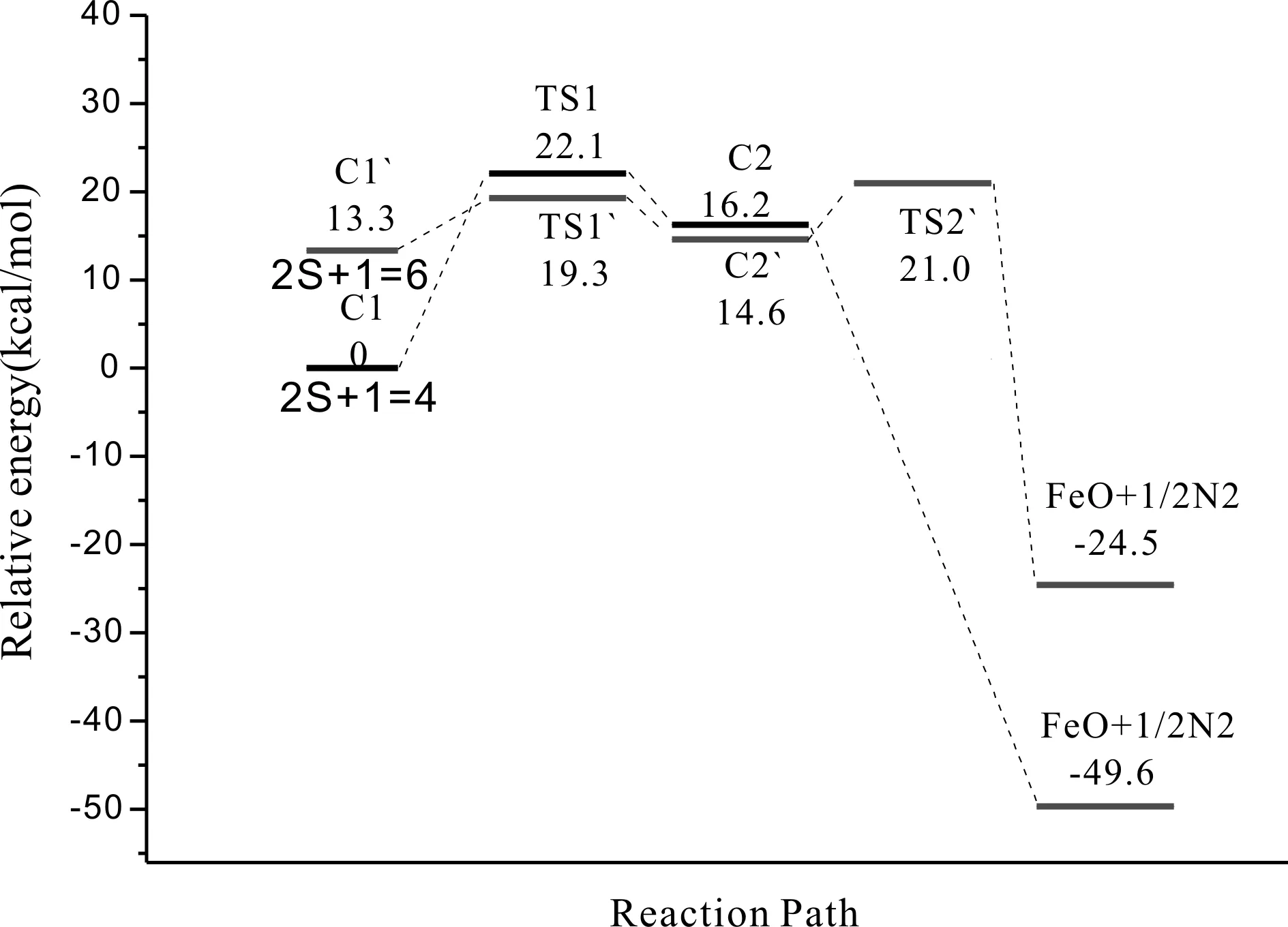
图8 Fe+NO在反应通道二中四重态和六重态的反应势能面图Fig. 8 Diagram of PESs for the reaction of Fe+NO on the quartet and sextet states in channel 2

图9 Fe+NO在反应通道三中四重态和六重态的反应势能面图Fig. 9 Diagram of PESs for the reaction of Fe+NO on the quartet and sextet states in channel 3
4 结 论
本文采用密度泛函理论的B3LYP方法对金属铁与NO的反应机理进行了分子水平的模拟研究.分别研究了三个反应通道上四、六重态反应势能面上的反应,结果表明金属铁原子能有效的把NO转化为FeO和N2,同时该体系在三个反应通道中进行时,都出现了势能交叉点,不仅能有效地降低整个反应过程中的势垒,还有利于反应动力学和热力学.本文为进一步研究金属铁有效催化脱除NO提供了一定的理论依据.
[1] Bacic Z, Miller R E. Molecular clusters: Structure and dynamics of weakly bound systems[J].J.Phys.Chem., 1996, 100(31): 12945.
[2] Castleman A W, Bowen K H. Clusters: structure, energetics, and dynamics of intermediate states of matter[J].J.Phys.Chem., 1996, 100(31): 12911.
[3] García-García A, Illán-Gómez M J, Linares-Solano A,etal. Potassium-containing briquetted coal for the reduction of NO[J].Fuel, 1997, 76(6): 499.
[4] Illan-Gomez M J, Raymundo-Pinero E. Catalytic NOxreduction by carbon supporting metals[J].Appl.Catal. B:Environmental, 1999, 20: 267.
[5] Yamashita H, Yamada H, Tomita A. Reaction of nitric oxide with metal-loaded carbon in the presence of oxygen[J].Appl.Catal., 1991, 78(1): L1.
[6] Ha Yhurst A N, Lawrence A D. The reduction of the nitrogen oxides NO and N2O to molecular nitrogen in the presence of iron, its oxides, and carbon monoxide in a hot fluidized bed [J].CombustFlame, 1997, 110 (3): 351.
[7] Zhong B J, Zhang H S. Experimental study of catalytic reduction of NO by lean coal chars[J].J.Eng.Thermophys., 2002, 23(2): 249 (in Chinese)[钟北京,张怀山. 贫煤焦催化还原NO 的实验研究[J]. 工程热物理学报, 2002, 23(2): 249]
[8] Su Y X, Deng W Y, SU A L. NO reduction by methane over oxides and the mechanism[J].J.Fuel.Chem.Tech., 2013, 41(9): 1129 (in Chinese)[苏亚欣, 邓文义, 苏阿龙. 甲烷在氧化铁表面还原NO的特性与反应机理研究[J]. 燃料化学学报, 2013, 41(9): 1129]
[9] Su Y X, Deng W Y, SU A L. NO reduction by methane on the surface of iron and iron oxides[J].J.Fuel.Chem.Tech., 2013, 41(11): 1393(in Chinese)[苏亚欣, 任立铭, 苏阿龙. 甲烷在金属铁及氧化铁表面还原NO的实验研究[J]. 燃料化学学报, 2013, 41(11): 1393]
[10] Su Y X, Su A L, Ren L M,etal. Effect of SO2on the reduction of NO by metheane over iron catalyst[J].J.Fuel.Chem.Tech., 2014, 42(3): 377 (in Chinese)[苏亚欣, 苏阿龙, 任立铭, 等. SO2对甲烷在金属铁表面还原NO的反应影响[J]. 燃料化学学报, 2014, 42(3): 377]
[11] Blagojevic V, Orlova G, Bohme D K.O-atom transport catalysis by atomic cations in the gas phase: Reduction of N2O by CO [J].J.Am.Chem.Soc., 2005, 127(10): 3545.
[12] Rondinelli F, Russo N, Toscano M. On the origin of the different performance of iron and manganese monocations in catalyzing the nitrous oxide reduction by carbon oxide[J].Inorg.Chem., 2007, 46(18): 7489.
[13] Chen D P, Kong C, Han Y X,etal. Theoretical study of catalytic oxidation cycles of CO with N2O by Fe+in gas phase[J].J.At.Mol.Phys., 2013, 30(4): 517(in Chinese)[陈东平, 孔超, 韩艳霞. 气相中Fe+催化CO与N2O循环反应的理论计算研究[J]. 原子与分子物理学报, 2013, 30(004): 517]
[14] Gan Y Z, Wang Y C, Jin Y Z. Theoretical study of the mechanism for the cycle reaction of N2O and CH4catalyzed by Fe+(6D) to yield CH3OH[J].Sci.ChinaSer. B, 2013, (006): 763 (in Chinese)[甘延珍, 王永成, 金燕子. Fe+(6D)催化N2O和CH4制取甲醇循环反应的理论探究[J]. 中国科学: 化学, 2013, (006): 763]
[15] Fiedler A, Iwata S. Variety of [Fe, N, O] isomers. A theoretical study[J].J.Phys.Chem. A, 1998, 102(20): 3618.
[16] Harvey J N. On the accuracy of density functional theory in transition metal chemistry[J].Annu.Rep.Prog.Chem.Sect. C:Phys.Chem., 2006, 102: 203.
[17] Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Phys.Rev. B, 1988, 37(2): 785.
[18] Sun Q, Li Z, Wang M,etal. Methane activation on Fe4cluster: A density functional theory study[J].Chem.Phys.Lett., 2012, 550: 41.
[19] Wang L, Wang G, Qu H,etal. Infrared photodissociation spectroscopy of iron nitrosyl cation complexes: Fe (NO)n+(n= 1-5)[J].J.Phys.Chem. A, 2014, 118(10): 1841.
[20] Sun Q, Kandalam A K, Wang Q,etal. Effect of Au coating on the magnetic and structural properties of Fe nanoclusters for use in biomedical applications: A density-functional theory study[J].Phys.Rev. B, 2006, 73(13): 134409.
[21] Xiao L, Wang L. Methane activation on Pt and Pt4: A density functional theory study[J].J.Phys.Chem. B, 2007, 111(7): 1657.
[22] Sun Q, Altarawneh M, Dlugogorski B Z,etal. Catalytic effect of CuO and other transition metal oxides in formation of dioxins: theoretical investigation of reaction between 2, 4, 5-trichlorophenol and CuO[J].Environ.Sci.Technol., 2007, 41(16): 5708.
[23] Danovich D, Shaik S. Spin-orbit coupling in the oxidative activation of HH by FeO+. Selection rules and reactivity effects[J].J.Am.Chem.Soc., 1997, 119(7): 1773.
[24] Van Koppen P A M, Bowers M T, Fisher E R,etal. Relative energetics of CH and CC bond activation of alkanes: reactions of Ni+and Fe+with propane on the lowest energy (adiabatic) potential energy surfaces[J].J.Amer.Chem.Soc., 1994, 116(9): 3780.
[25] Schröder D, Shaik S, Schwarz H. Two-State reactivity as a new concept in organometallic chemistry[J].Acc.Chem.Res., 2000, 33(3): 139.
Density functional theory study of the reaction of iron atom with NO
QI Yue-Zhou, SU Ya-Xin
(School of Environmental Science and Engineering, Donghua University, Shanghai 201620, China)
Density functional theory (DFT) calculations have been carried out to study the micro-mechanism for reaction of iron atom with NO. The geometry optimizations of reactants, transition states, intermediates and products of the reactions of sextet and quartet states were completely optimized, and all the transition states were verified by the vibrational analysis and the intrinsic reaction coordinate calculations. Then the potential energy surface (PES) were obtained and crossing points were investigated. Results showed that the reaction of iron atom with NO was a typical two-state reaction(TSR). One crossing point appeared in the reaction channels 1, Two crossing points appeared in the reaction channels 2, while three crossing points appeared in the reaction channels 3 between the quartet and the sextet potential energy surfaces, which would effectively reduce the activation energy and increase the release of reaction heat, play a significant and beneficial role in the kinetic and thermodynamic aspects of this catalytic reaction.
Transition metal atom; Quantum chemistry; Micro-mechanism of reaction; Crossing point
103969/j.issn.1000-0364.2015.12.005
2014-10-28
国家自然科学基金(51278095)
戚越舟(1989—), 男,硕士研究生,主要研究烟气脱硝及量子化学模型.
苏亚欣. E-mail: suyx@dhu.edu.cn
O641
A
1000-0364(2015)06-0924-07
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
大学物理(2022年1期)2022-01-13
World Journal of Psychiatry(2020年4期)2020-07-11
发明与创新(2020年11期)2020-03-31
小学生学习指导(中年级)(2019年3期)2019-04-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
电子制作(2017年19期)2017-02-02
科技视界(2016年24期)2016-10-11
中华建设科技(2014年6期)2014-08-27
