
Rh的芳基化合物与含氮芳基卤代物交叉偶联反应机理研究
2015-03-22 10:27王晓岚徐铭瑶胡庆仟李来才
原子与分子物理学报 2015年6期
王晓岚, 徐铭瑶, 胡庆仟, 李来才
(四川师范大学化学与材料学院, 成都 610066)
Rh的芳基化合物与含氮芳基卤代物交叉偶联反应机理研究
王晓岚, 徐铭瑶, 胡庆仟, 李来才
(四川师范大学化学与材料学院, 成都 610066)
利用密度泛函理论(DFT)研究了Rh的芳基化合物与含氮芳基卤代物交叉偶联过程催化循环的微观反应机理.在B3LYP/6-31+G(d) 基组水平上(Rh、I采用了赝势基组LanL2DZ)优化了反应过程中所有化合物的几何构型并计算了频率,通过能量、频率和振动方式确定了中间体和过渡态的真实性.此外,在同等基组水平上还运用了分子中的原子理论讨论了成键临界点的电荷密度的变化,运用了自然键轨道理论讨论了键的性质与轨道间的相互作用.为了提高计算精度, 在6-311++G(d,p) 基组水平上计算了反应机理中所有物质在气相及溶剂化下的单点能, 得到与 6-31+G(d) 基组计算相同的结论.结论表明Rh(Ⅰ)起到了有效的催化作用,且计算所得结论与实验结果相符合.
密度泛函理论; 芳香化合物; 含氮芳基卤代物; Rh(Ⅰ)催化; 交叉偶联反应; 微观反应机理
1 引 言
C—C键形成的偶联反应在有机合成领域中是重要的研究内容之一[1-3].高效高选择性的C—C偶联反应的新反应和新途径能够大大提高目标产物的合成效率,对有机合成化学的应用和发展具有很大的应用价值和理论意义.构造C—C键成键的偶联反应效率由于有机合成化学中的周环反应[4]和过渡金属催化的偶联反应的发展而得到极大提高[5,6].卤素原子因为可以改变分子的空间结构和亲水亲油性,从而影响整个化合物分子的物理属性、化学性质和生物活性而在有机合成中占有一个重要的位置.同时卤代芳烃化合物中的卤素官能团能通过亲核加成、亲核取代、氧化加成等反应转化为其他官能团,进而改变化合物的化学性质,达到预期合成效果.因此作为化学原料以及医药中间体中重要的一份子,芳基卤代物被广泛使用[7-10].由Rh的配合物来作催化剂的有机卤代物和金属有机化合物的交叉偶联反应的反应机理的研究比较少见[11-14].Rh的配合物作为催化剂在有机合成反应具有可选择性高,活性很好,所需反应条件比较温和等特点,已经广泛运用于精细工业、石油工业、医药化工等领域[15-21].但作为一种稀有金属,Rh的回收循环再利用很重要.Ryo Shintani等已经在实验室里运用Rh配合物作为催化剂完成了多组份有机物的偶联反应[22].因此本文想通过研究Rh(Ⅰ)催化下的Rh芳基化合物与含氮芳基卤代物偶联反应的催化循环的理论研究,分析其最可能实现的反应路径,更清晰得了解其微观反应机理,为类似的后续实验起到一定的指导作用.
2 计算方法
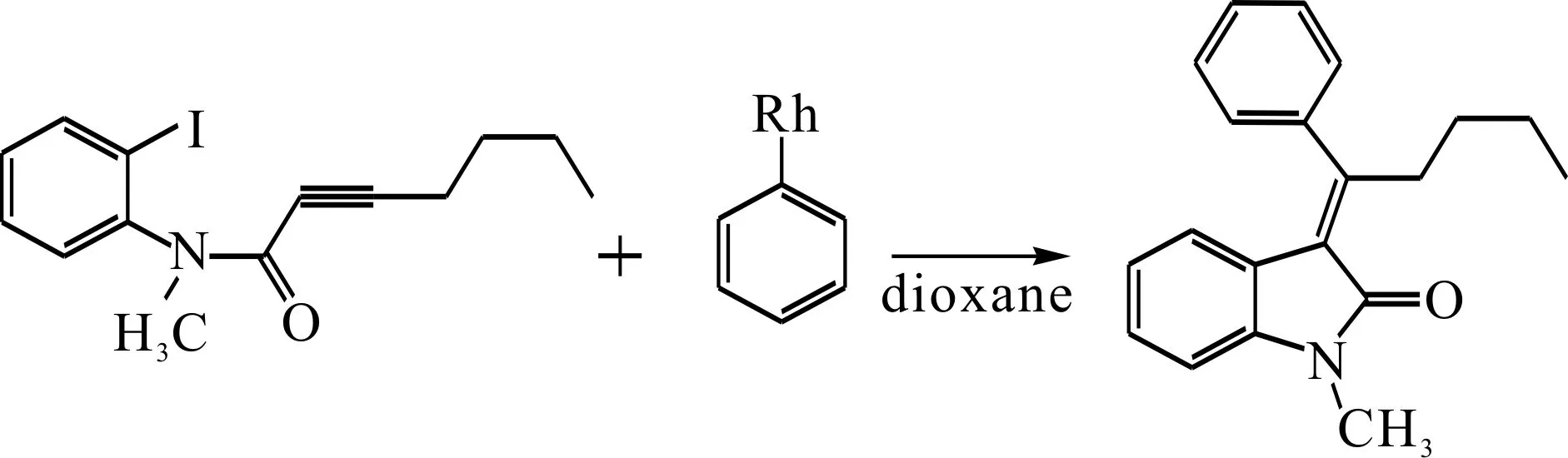
本文对Rh(Ⅰ)催化下的Rh芳基化合物与含氮芳基卤代物偶联反应的催化循环过程中所有的反应物、中间体、过渡态和产物利用密度泛函理论的B3LYP方法,在6-31+G (d) 基组水平上(Rh和I采用了赝势基组LanL2DZ)进行了结构优化,并在相同基组水平下对各构型进行了频率计算.反应公式如下:
运用自然键轨道(NBO)方法分析了所有物质的轨道间相互作用[23].使用自洽反应场(SCRF)极化连续模型(IEF-PCM)模拟实验所使用的二氧六环(dioxane)溶剂效应, 采用6-311++G (d, p) 基组水平对反应机理中各化合物均进行了全参数优化,并使用相同基组计算了气相条件下的单点能.为了进一步的了解成键性质分析成键特征,采用AIM2000的程序包计算了对应的所有成键临界点(BCP)和成环临界点(RCP) 电荷密度,显示分子中原子的属性[24].所有计算均采用Gaussian09程序完成[25].
3 反应机理和能量分析
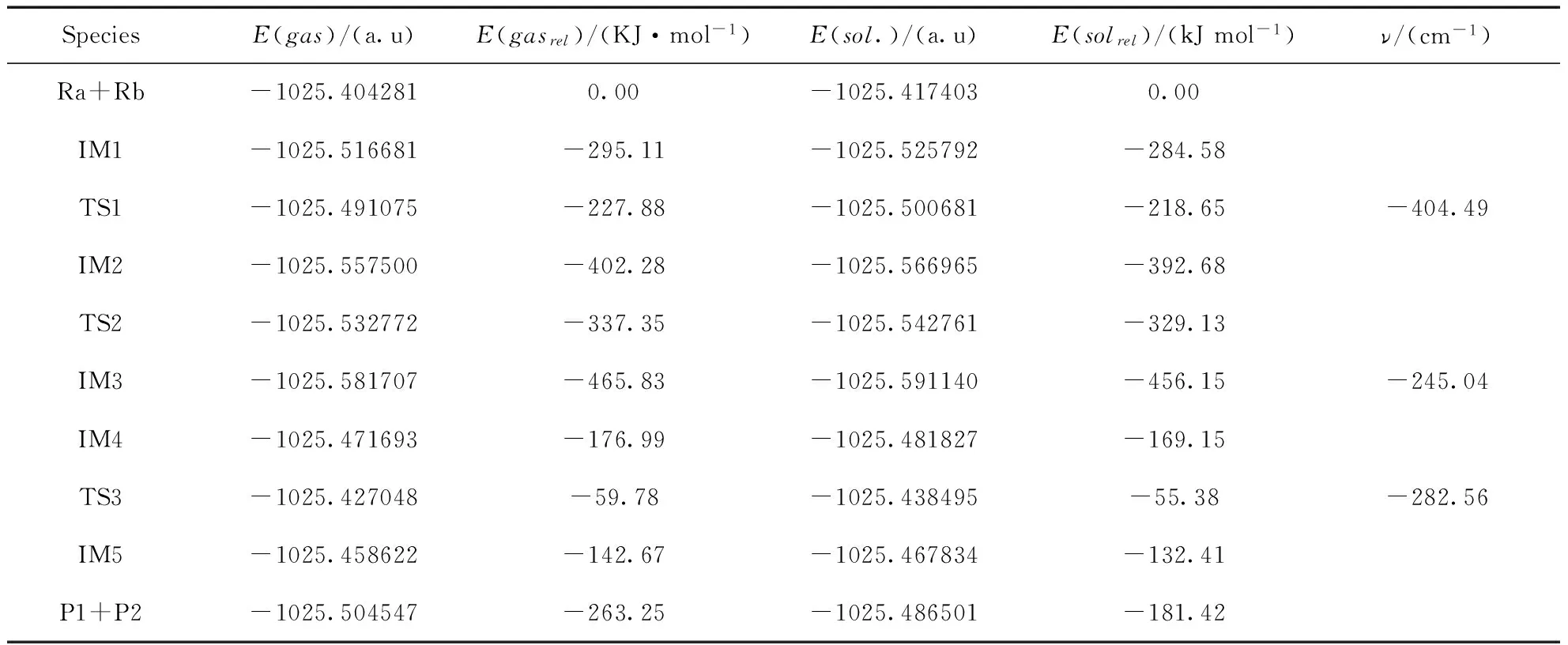
实验报道中, 在反应条件筛选时使用的溶剂为二氧六环(dioxane), 因此在进行反应机理的理论研究时选用与实验一致的dioxane作为溶剂.对在6-31+G(d) 基组下优化后的反应物、中间体、过渡态以及生成物的构型使用大基组6-311++G(d,p) 进行了单点能计算,气相和溶剂化条件下通过计算所得能量(E)、相对能量(Erel)及过渡态频率数值均已列于表1,能量(E)已包括零点能(ZPE)修正.
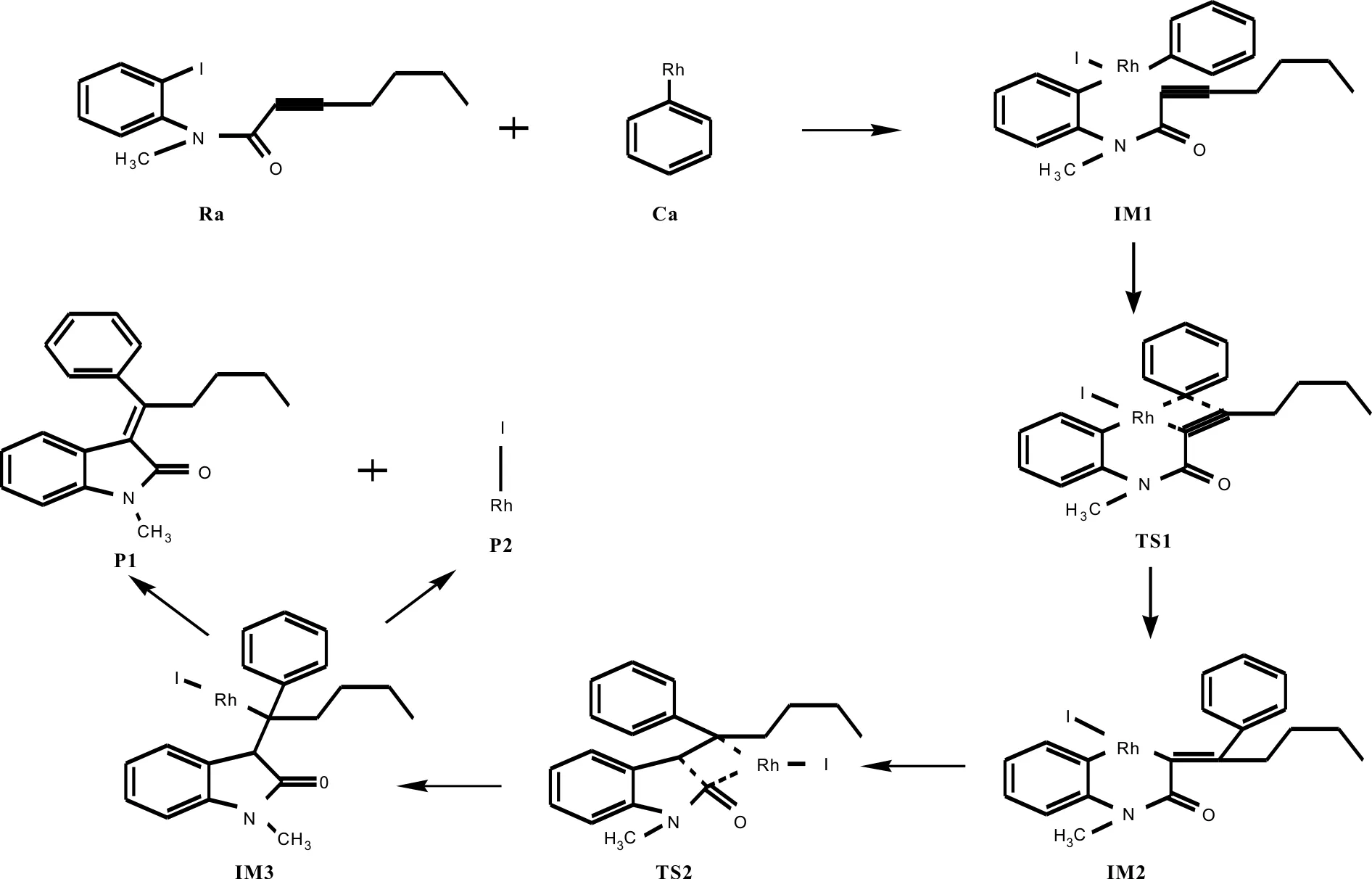
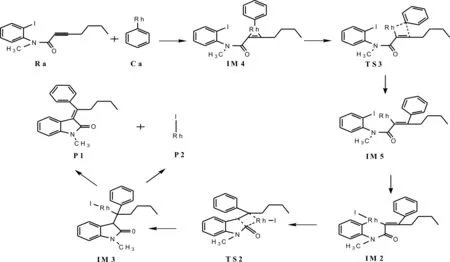
Scheme 1、2即为此偶联反应可能存在的计算模拟过程图,过程中还存在一个含炔烃的芳烃偶氮的芳基化交叉偶联反应过程.将本文计算模拟出的可能的反应通道scheme1、2与文献相比较[22],可以看出本文与Ryo Shintani所推断的反应过程相一致.整个偶联反应过程中所涉及的所有反应、中间体、过渡态及产物的构型全部列于图1及图3中.
3.1 反应通道一
在推断的反应通道一中,具体流程图见Scheme1.Rh插入卤素之间发生氧化加成反应,发生金属转移,促使芳基上的C与炔基上的C发生交叉偶联,金属继续发生转移,最后通过还原消除反应,催化剂顺利脱落.
反应物Ra与Rh的化合物Ca发生反应后,Ca中的Rh原子插入反应物的苯环和I原子之间生成中间体IM1,体系的总能量降低了295.11 KJ/mol.IM1构型是稳定存在,催化剂的插入过程较易完成.
IM1经由过渡态TS1生成IM2,这是一个分子内的插入反应过程. 反应活化能为67.23 KJ/mol,是反应通道一的速控步骤.与IM1相比较,TS1中的Rh-C(3)键长增大了0.098 Å,Rh原子和C(1)原子之间的距离由2.180 Å减少至1.988 Å,C(2)原子和C(3)原子的距离由之前的2.214 Å减少至2.122 Å.Rh原子和C(3)原子有相互远离的趋势,而Rh-C(1)以及C(2)-C(3)已经微弱成键.TS1有且仅有一个虚频为-404.49 cm-1.通过NBO分析,得到Rh-C(1) 的双中心成键轨道上电子占据数为1.77815个,双中心反键轨道电子占据数为0.34350个,也说明Rh-C(1) 已微弱成键.在中间体IM2中,C(2)-C(3)之间的键长和电荷密度分别为1.484 Å和0.269 a.u..C(2)与C(3)原子间的键长明显缩短,电荷密度增大,C(2)-C(3)成键.Rh原子与C(3)原子距离增大,Rh-C(3)键断开.通过TS1,实现了由Rh催化的芳基化交叉偶联反应过程.接着IM2经过一个过渡态TS2生成中间体IM3,这是一个Rh原子转移促使C-C偶联发生的过程.IM2至TS2的反应活化能为64.93 KJ/mol.TS2的虚频唯一,值为-245.04 cm-1,说明了此过渡态的真实存在.与IM2相比较,TS2中的C(1)原子与C(4)原子的键长由2.753 Å缩短至1.484 Å,且存在一个0.296 a.u.的键鞍点电荷密度,说明C(1)-C(4)已逐渐成键,达到C-C偶联的效果.而通过对TS2的NBO分析,我们可以发现C(1)-C(4)的双中心成键轨道上电子占据数为1.96634个,双中心反键轨道电子占据数为0.04119个,也说明C(1)-C(4)已微弱成键.从TS2到IM3,体系能量降低了128.48 KJ/mol.与TS2相比较,C(1)-C(4)键长更短,成键更稳定;Rh原子与C(2)原子之间的距离由3.022 Å缩短至2.116 Å,逐渐成键.也就是说在C(1)与C(4)原子不断靠拢的同时,Rh与I原子不断朝C(2)方向靠近.通过对IM3的NBO分析,C(1)-C(4)键主要是SP-SP杂化轨道形成的σ键,电子占据数为1.96511个,由金属Rh催化的C(1)-C(4)偶联过程完成.
表1 反应各驻点的能量G(a.u.)、相对能量Grel(KJ·mol-1)及各过渡态频率v(cm-1)
Table 1 The energies (E/a.u.), relative energies (Erel/KJ·mol-1) and frequencies (v/cm-1) of the compounds
SpeciesE(gas)/(a.u)E(gasrel)/(KJ·mol-1)E(sol.)/(a.u)E(solrel)/(kJmol-1)ν/(cm-1)Ra+Rb-1025.4042810.00-1025.4174030.00IM1-1025.516681-295.11-1025.525792-284.58TS1-1025.491075-227.88-1025.500681-218.65-404.49IM2-1025.557500-402.28-1025.566965-392.68TS2-1025.532772-337.35-1025.542761-329.13IM3-1025.581707-465.83-1025.591140-456.15-245.04IM4-1025.471693-176.99-1025.481827-169.15TS3-1025.427048-59.78-1025.438495-55.38-282.56IM5-1025.458622-142.67-1025.467834-132.41P1+P2-1025.504547-263.25-1025.486501-181.42
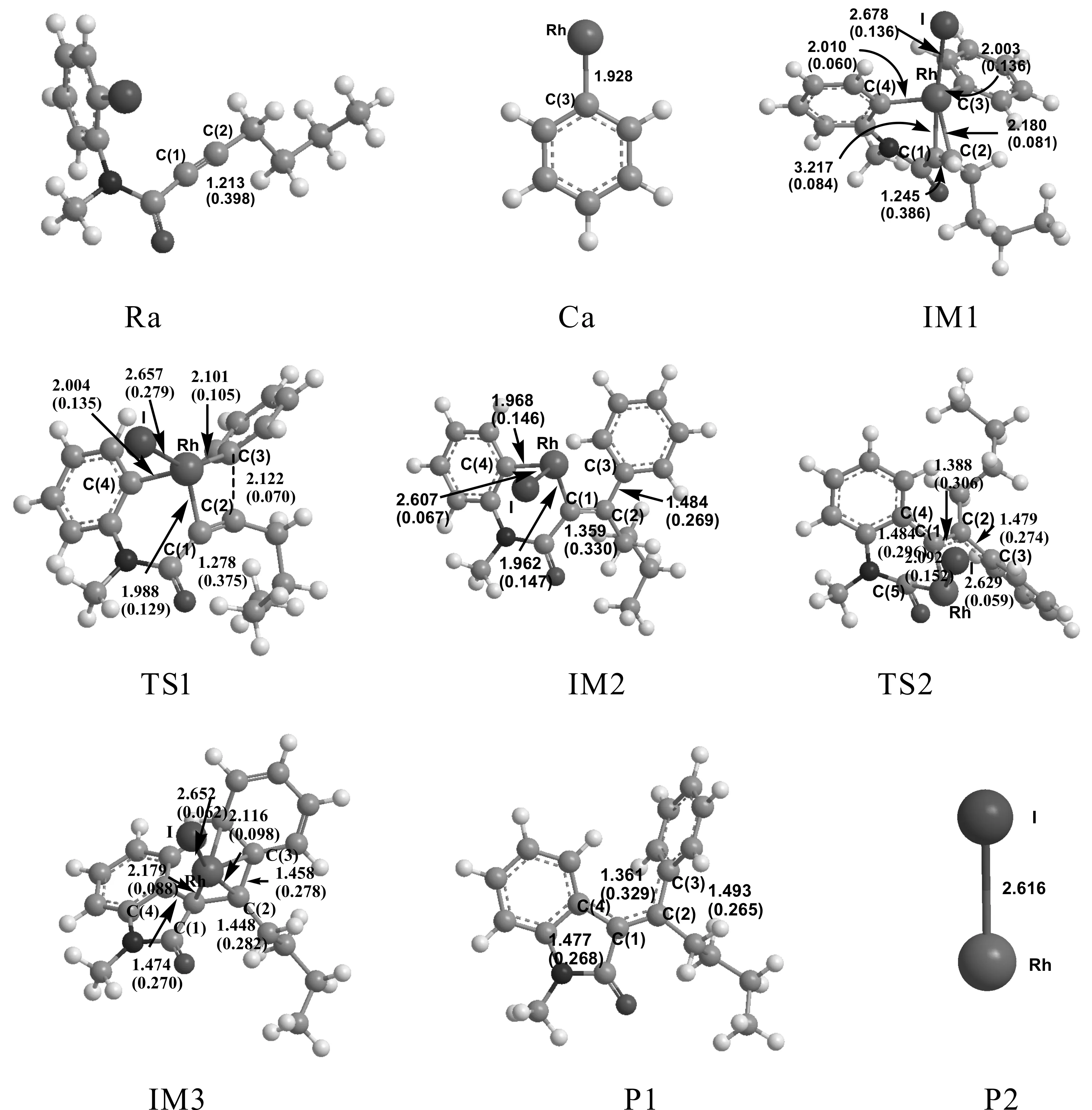
图1 反应通道一中涉及的化合物的几何优化构型:键长单位为Å,电荷密度单位为a.u.Fig. 1 Optimized structures of the compounds of the channel 1:bond length in angstrom units, and charge density at the bond-forming critical point in atomic units
Scheme 1 反应通道一的微观流程图Scheme 1 The mechanism of the reaction channel 1
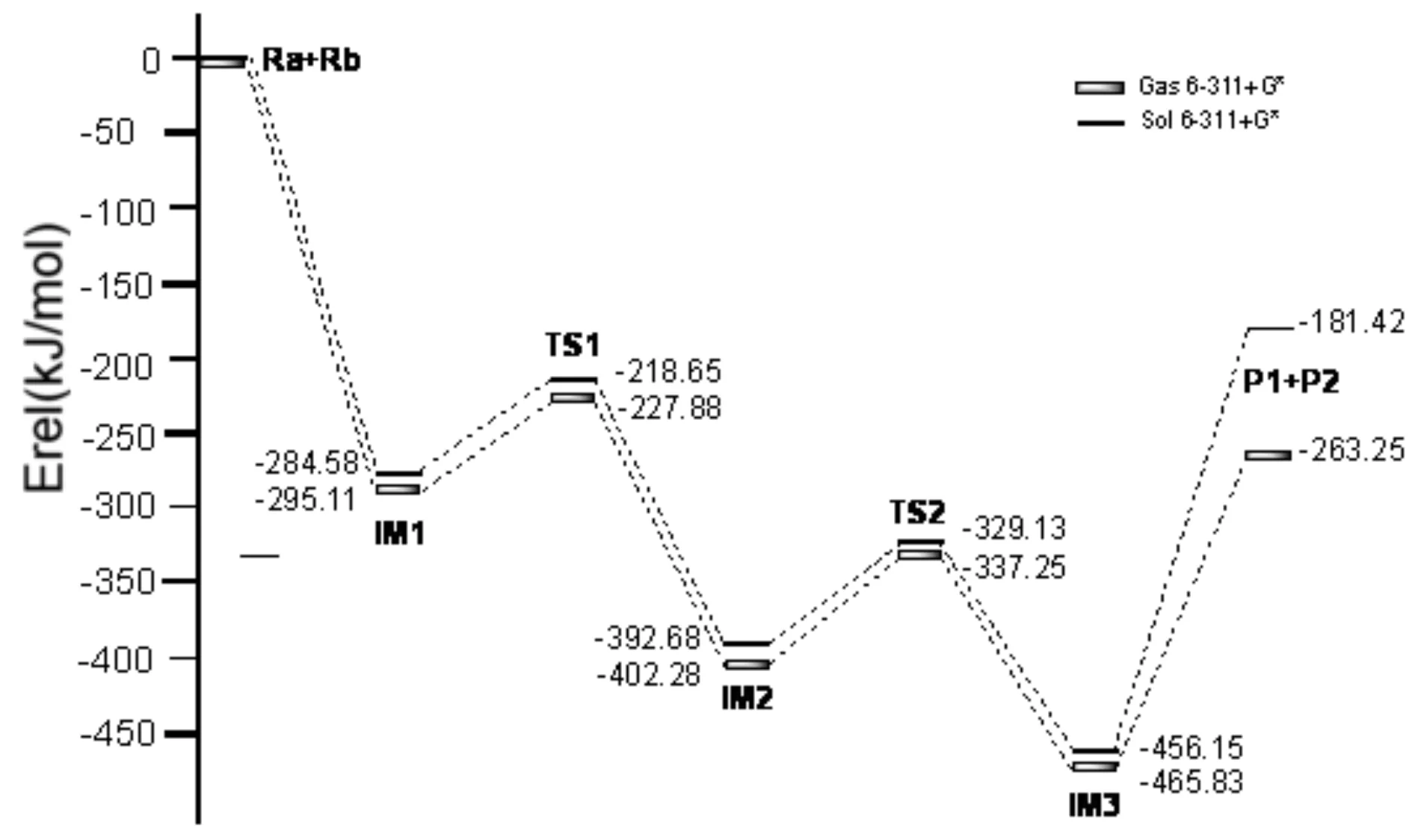
图2 反应通道一的能级图Fig. 2 Schematic of energy levels in the channel 1 of the reaction
接下来IM3脱去催化剂Rh,生成产物P1和P2,完成整个催化过程,开始新一轮的催化循环.产物P1中C(1)-C(4)之间的键长为1.477 Å,电荷密度为0.268 a.u.; C(1)-C(2)键长为1.361 Å,电荷密度为0.329 a.u.;C(2)-C(3)之间的键长和电荷密度分别为1.493 Å和0.265 a.u..最终,实现了产物的芳基化交叉偶联以及C-C偶联反应过程.与实验结果相一致.
Rh(Ⅰ)催化下的Rh芳基化合物与含氮芳基卤代物偶联反应的催化循环过程一可能为:Ra+Ca →IM1 →TS1 →IM2 →TS2 →IM3→ P1+P2.反应通道一的速控步骤为IM1 →TS1 →IM2,反应活化能为67.23 KJ/mol.结合表1所列在相同基组水平上溶剂化条件下理论计算所得的能量数据,可知能量变化趋势和气相条件下是一致的.
3.2 反应通道二
因为Rh配合物容易与各种不饱和键发生加成,因此第二个可能存在的反应通道初始时是由Rh的芳基化合物直接插入反应物中不饱和的C-C键,反应具体流程图见Scheme 2.
即反应物Ra与Rh化合物Ca发生反应后生成中间体IM4,体系能量降低284.58 KJ/mol.说明IM4的构型稳定存在,插入催化剂过程较容易完成.催化剂的插入过程完成,生成一个很稳定的Rh的烯基化合物.
接着中间体IM4经由过渡态TS3生成另一个稳定的中间体IM5,这是一个分子内的氧化加成反应过程.IM4至TS3的反应活化能为117.21 KJ/mol,是反应通道二的速控步骤.由过渡态TS3完成苯基的交叉偶联到稳定状态IM5,体系能量降低了82.89 KJ/mol.TS3的虚频唯一,值为-282.56 cm-1,说明了此过渡态的真实存在.与IM4相比较,C(2)原子与C(3)原子的键长缩短,存在一个0.113a.u.的键鞍点电荷密度,说明C(2)-C(3)已逐渐偶联.对TS3的NBO分析发现C(2)-C(3)的双中心成键轨道上电子占据数为1.73685个,双中心反键轨道电子占据数为0.25796个,该键相对能量为-0.46351 a.u.,也说明C(2)-C(3)已有成键趋势.IM5与TS3相比较,C(2)-C(3)键的键长更短,成键更稳定,说明IM5是一个稳定存在的中间体.在Rh催化作用下,电子的跃迁和迁移导致苯环靠拢向炔基的另一端,完成芳基化的交叉偶联反应过程.通过对IM5的NBO分析,C(2)-C(3)键主要是SP-SP杂化轨道形成的σ键,双中心成键轨道上电子占据数为1.96460个,双中心反键轨道电子占据数为0.04065个,该键相对能量为-0.62967 a.u..C(2)-C(3)稳定成键,Rh-C(3)断开,由金属Rh催化的C(2)-C(3)偶联过程完成.接下来的反应步骤与反应通道一反应步骤趋于相同,IM5通过一个分子内的插入反应形成IM2,Rh原子直接插入卤素与C原子之间形成一个新的Rh化合物.系统能量降低了259.61 KJ/mol,反应较易进行.IM2经过TS2生成IM3,IM3脱去催化剂Rh,生成产物P1和P2,完成催化过程,开始新一轮的催化循环.最终实现了产物的芳基化交叉偶联以及C-C偶联反应过程.与实验结果相一致.Rh(Ⅰ)催化下的Rh芳基化合物与含氮芳基卤代物偶联反应的催化循环过程二可能为:Ra+Ca →IM4 →TS3 →IM5→IM2 →TS2 →IM3→ P1+P2.整个反应的速控步骤为IM4 →TS3 →IM5,反应活化能为117.21 KJ/mol.结合表1所列在相同基组水平上溶剂化条件下理论计算所得的能量数据,可知能量变化趋势和气相条件下是一致的.与反应通道二相比较,通道一的活化能更低,更容易发生,更容易清楚地体现整个偶联反应的微观过程.
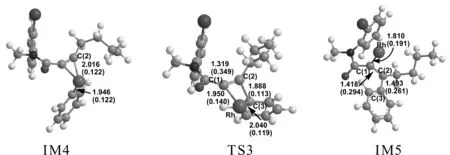
图3 反应通道二中涉及的化合物的几何优化构型:键长单位为Å,电荷密度单位为a.u.Fig. 3 Optimized structures of the compounds of the channel 2:bond length in angstrom units, and charge density at the bond-forming critical point in atomic units
Scheme 2 反应通道二的微观流程图Scheme 2 The mechanism of the reaction channel 2
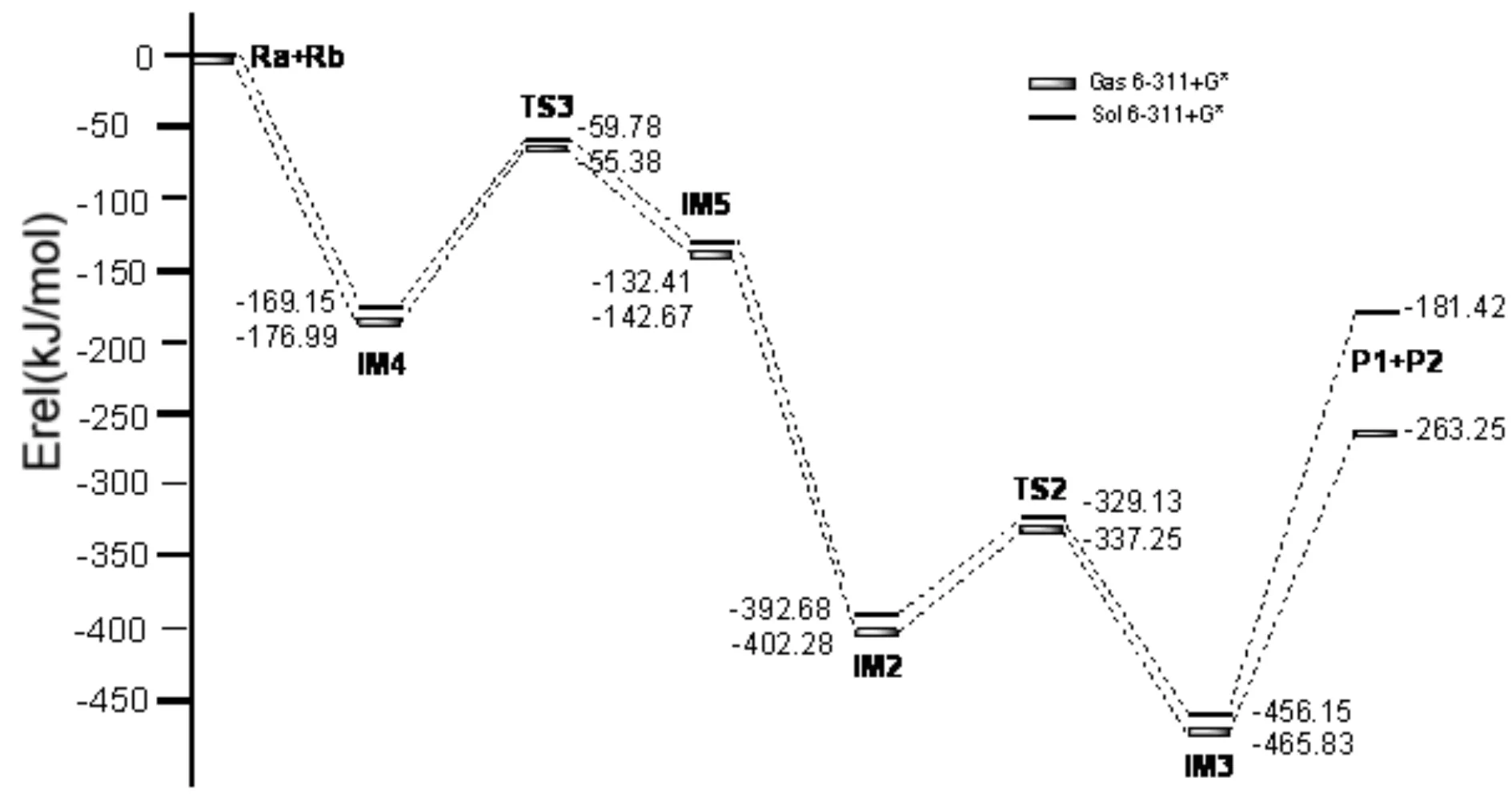
图4 反应通道二的能级图Fig. 4 Schematic of energy levels in the channel 2 of the reaction
4 结 论
本文采用密度泛函理论(DFT)的B3LYP方法,在6-31+G(d) 基组水平上(Rh和I采用了赝势基组LanL2DZ)研究了Rh芳基化合物与含氮芳基卤代物偶联反应的催化循环过程中的两种可能存在的反应通道的微观反应机理.同时使用自洽反应场(SCRF)极化连续模型(IEF-PCM)模拟实验所使用的二氧六环(dioxane)溶剂效应, 采用6-311++G(d,p)基组水平对反应机理中各化合物均进行了全参数优化,并使用相同基组计算了气相条件下的单点能.全文分析了此偶联反应的两种可能存在的反应通道,结论如下:(1)Rh(Ⅰ)在偶联反应中是一种有效的催化剂.(2)Rh配合物作为催化剂的偶联反应依然和其他的偶联反应一样遵循氧化加成—金属配体转移—还原消除,催化剂进入下一个循环这样的合成步骤.(3)在溶剂化条件下反应能量变化趋势和气相条件下是一致的.(4)计算模拟出的微观机理过程及所得结论与Ryo Shintani等的实验推断过程及实验结论相符合.
[1] Zhang W W, Dai Z Q, Zhang Z Y,etal. Progress in transition-metal-catalyzed cross-coupling reaction of Grignard reagent with halides [J].ChemicalWorld, 2011, 5: 314 (in Chinese) [张炜炜, 戴志群, 张智勇, 等. 过渡金属催化下的格氏试剂与卤代烃的偶联反应研究进展[J]. 化学世界, 2011, 5: 314]
[2] Corey E J, Cheng X M.Thelogicofchemicalsynthesis[M]. New York: John Wiley & Sons, 1989.
[3] Guo X W, Li Z P, Li C J. Cross-dehydrogenative-coupling(CDC)reaction [J].Process.Chem., 2010, 22: 1434 (in Chinese) [郭兴伟,李志平,李朝军. 交叉脱氢偶联反应[J]. 化学进展, 2010, 22: 1434]
[4] Fleming I.Pericyclicreactions[M]. New York: Oxford University Press, 1999.
[5] Crabtree R H.Theorganometallicchemistryoftransitionmetals,4thed[M]. New York: Wiley Interscience, 2005.
[6] Tsuji J.Transitionmetalreagentsandcatalysts:innovationsinorganicsynthesis[M]. Chichester, UK: Wiley, 2002.
[7] Novák Z, Szabó A, Répási J,etal. Sonogashira coupling of aryl halides catalyzed by palladium on charcoal [J].J.Org.Chem., 2003, 68 (8): 3327.
[8] Hanamoto T, Kobayashi T. Cross-coupling reactions of (1-fluorovinyl) methydiphenylsilane1 with aryl halides and aryl triflates [J].J.Org.Chem., 2003, 68 (16): 6354.
[9] Everson D A, Shrestha R, Weix D J. Nickel-catalyzed reductive cross-coupling of aryl halides with alkyl halides [J].J.Am.Chem.Soc., 2010, 132 (3): 920.
[10] Wager K M, Daniels M H. Palladium-catalyzed cross-coupling of benzyl thioacetates and aryl halides [J].Org.Lett., 2011, 13(15): 4052.
[11] Hossain K M, Takagi K. Novel Rh(I)-catalyzed reaction of arylzinc compounds with methyl halides [J].Chem.Lett., 1999, 28: 1241.
[12] Takahashi H, Hossin K M, Nishihara Y,etal. Synthesis of functionalized benzylsilanes from arylzinc compounds and (Iodomethyl) trimethylsilane by means of a novel Rh catalysis [J].Org.Chem., 2006, 71: 671.
[13] Takahashi H, Inagaki S, Nishihara Y,etal. Novel Rh catalysis in cross-coupling between alkyl halides and arylzinc compounds possessing ortho-COX (X = OR, NMe2, or Ph) groups [J].Org.Let., 2006, 8: 3037.
[14] Yasui H, Mizutani K, Yorimitsu H,etal. Cobalt- and rhodium-catalyzed cross-coupling reaction of allylic ethers and halides with organometallic reagents [J].TetrahedronAsy., 2006, 62: 1410.
[15] Han Y F, Yang D Q. Progress of Rhodium-catalyzed asymmetric hydrogenation [J].ChemicalIndustryTimes, 2004, 18: 1 (in Chinese) [韩英峰, 扬定乔. 铑催化不对称氢化反应的研究进展[J].化工时刊. 2004, 18: 1]
[16] Joel M, Hawkins T J N. Asymmetric catalysis in the pharmaceutical industry [J].AngewChem.Int.Ed., 2004, 43: 3224.
[17] Jerphagon T, Renaud J, Bruneau C. Chrial monodentate phosphorus ligands for rhodium -catalyzed asymmetric hydrogenation [J].TetrtahedronAsy., 2004, 15: 2101.
[18] Dieguez M, Pamies O, Claver C. Recent advances in Rh-catalyzed asymmetric hydroformformylation using phosphite ligands [J].TetrtahedronAsy., 2004, 15: 2113.
[19] Chen B, Dingerdissen U, Krauter J G E,etal. New developments in hydrogenation catalysis particularly in synthesis of fine and intermediate chemicals [J].AppliedCatalyst., 2005, 280: 17.
[20] Blaser H U, Pugin B, Spindler F. Progress in enantioselective catalysis assessed from an industrial point of view [J].J.Mol.Catal. A:Chem., 2005, 231: 1.
[21] Xiong X D, Wang S G. Application of Platinum-group metals in asymmetric catalysis [J].ChineseJournalofRareMetals, 2005, 29(3): 363 (in Chinese) [熊晓东, 王胜国. 铂族金属在不对称催化中的应用[J]. 稀有金属, 2005, 29(3): 363]
[22] Shintani R, Yamagami T, Hayashi T. Rhodium-catalyzed multicomponent-coupling reactions involving a carborhodation cross-coupling sequence [J].Org.Lett., 2006, 8: 4799.
[23] Reed A E, Weinhold F, Curtiss L A,etal. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint [J].J.Chem.Rev., 1988, 88 (6): 899.
[24] Bader R W F.Atomsinmolecules.Aquantumtheory[M]. Oxford: Oxford University Press, 1990.
[25] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 03, Rev. A7. Wallingford Gaussian, Inc., CT: 2009.
Theoretical investigation on the cross-coupling reaction mechanism of rhodium aromatic compounds with nitrogen-containing aryl halides
WANG Xiao-Lan, XU Ming-Yao, HU Qing-Qian, LI Lai-Cai
(College of Chemistry and Material Science, Sichuan Normal University, Chengdu 610066, China)
The cross-coupling reaction mechanism of rhodium aromatic compounds with nitrogen-containing aryl halides has been investigated by density functional theory (DFT). The geometries and the frequencies of reactants, intermediates, transition states, and products have been calculated at the B3LYP/6-31+G(d) level, and the LanL2DZ basis has been used as the extrabasis. The vibration analysis demonstrates that the authenticity of transition states, and the reaction processes are confirmed by the changes of charge density at bond-forming critical point analyzed by the atoms in molecules theory. In addition, the nature bond orbital has been used to discuss the bond nature and orbital interactions at the same level. Meanwhile, the single point energies of the reaction process in gas and solvent at 6-311++G(d,p) level have been individually investigated with higher precision. The results indicate that the reaction mechanism and the change trend of correspondence energy at two different levels are consistent. The result of the theory study agrees with the experimental data, it indicates that the Rh(Ⅰ) is an effective catalyst in this reaction.
Density functional theory; Aromatic compounds; Nitrogen-containing aryl halides; Rh(Ⅰ)-catalyze; Cross- coupling reaction; Reaction mechanism
103969/j.issn.1000-0364.2015.12.001
2014-11-06
四川师范大学重点资助项目
王晓岚(1984—),女,助教,硕士,主要从事应用量子化学研究.E-mail: 29757739@qq.com
李来才. E-mail: lilcmail@163.com
O561.4
A
1000-0364(2015)06-0903-07
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
北京航空航天大学学报(2022年5期)2022-06-06
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
