
几种N-取代-3-羟基-2-吡啶硫酮Be配合物的结构与光谱性质
2015-03-22 10:27黄荣谊吴根华
原子与分子物理学报 2015年6期
黄荣谊, 吴根华
(安庆师范学院化学化工学院功能配合物安徽省重点实验室, 安庆 246011)
几种N-取代-3-羟基-2-吡啶硫酮Be配合物的结构与光谱性质
黄荣谊, 吴根华
(安庆师范学院化学化工学院功能配合物安徽省重点实验室, 安庆 246011)
在B3LYP/6-31+G(d)级别水平上对4种Be配合物的几何结构进行了全优化,并探讨了烷基取代基对其分子的几何结构和电子结构等方面的影响.采用TD-B3LYP方法在同样级别水平上研究了各配合物的电子吸收光谱,分析了光谱的变化规律.上述计算结果表明,随烷基取代基团给电子能力的增强,前线分子轨道能级升高、能隙增大、最大吸收波长发生蓝移,且最大吸收波长的跃迁类型为配体内的π→π*跃迁.
Be配合物; 电子结构; 电子光谱; 含时密度泛函理论
1 引 言
近年来,功能配合物光学材料由于具有较好的热稳定性、较高荧光效率和易成膜性等特性已成为最有应用前景的一类光致/电致发光材料[1]. 配合物发光材料的光物理、光化学性能的研究已是世界各国科学家们的关注热点[2].迄今为止,绝大多数配合物光致/电致发光材料(稀土发光材料除外)所包含的金属离子多属于第Ⅱ、第Ⅲ族.如第Ⅲ族中配位数为6的Al3+和4的B3+以及第Ⅱ族中配位数为4的Be2+和Zn2+应用较为广泛[3]. 有机配体对配合物发光材料的光物理和光化学性能起着决定作用[4]. N-取代-3-羟基-2-吡啶硫酮是一类具有O、S双功能配位原子的螯合配体,易于与金属离子结合形成性质优良的功能配合物材料[5-7]. 目前,在理论上还未见含该类配体的Be的配合物的研究报道. 另外,在有机配体上引入特定给/吸电子基团,以提高分子平面性、增大其共轭程度、调节其前线分子轨道能隙、改善其电荷转移能力被认为是开发新型配合物发光材料的行之有效方法. 因此,本文以3-羟基-2-吡啶硫酮Be配合物为基本计算模型,引入一系列的烷基取代基(如甲基、乙基和丙基),从理论上探讨了取代基对其结构和光谱性质的影响,以期为实验研究提供一定的理论信息.
2 计算模型和方法
本文运用Gaussian 09 D.01程序软件[8],采用DFT/B3LYP方法,在6-31+g(d)基组下,对4种N-取代-3-羟基-2-吡啶硫酮Be配合物(图1)基态结构进行全优化和振动分析,振动分析计算无虚频. 利用AIM理论对各稳定几何构型进行了电子密度拓扑分析.另外,采用TD-DFT/6-31+g(d) 级别方法计算了其电子吸收光谱.
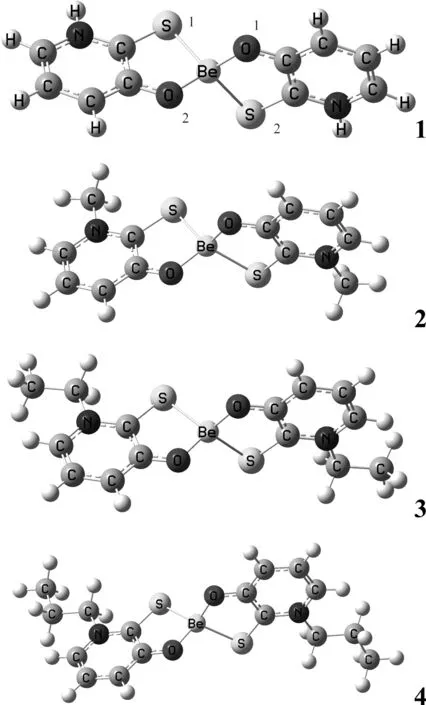
图1 配合物1-4的分子结构模型Fig. 1 Calculation models of complexes 1-4
3 结果与讨论
3.1 基态几何构型
计算的4种配合物的稳定几何构型如图1所示. 分子点群均为C2群,电子态均为1A.表1给出了4种配合物的稳定构型的部分优化几何参数. Be—O键键长在0.15825~0.15838 nm之间,Be—S键键长在0.21973~0.22110 nm之间,与文献报道的键长相近. 比较4种配合物的结构参数,可以看出烷基的引入对配合物结构产生了一定的影响,烷基给电子能力的增强,使得Be—O键和Be—S键的键长有规律的变短,但变化幅度较小. 同时Be原子和配位原子之间的键角在烷基的影响下也发生了些微变化. 4种配合物的吡啶环上的C—C键和C—N键以及环外的C—O键、C—S键键长均介于相应键的单、双键之间,表明吡啶环和S、O原子间共轭性较强. 因此,取代基的引入对3-羟基-2吡啶硫酮Be配合物的结构和性质将产生一定影响.
表1 在B3LYP/6-31+G(d)水平上优化的基态部分几何参数(键长: ×10-1nm;键角: °)
Table 1 Selected geometry parameters for the ground states optimized at B3LYP/6-31G(d) level (bond length: ×10-1nm; bond angle: °)
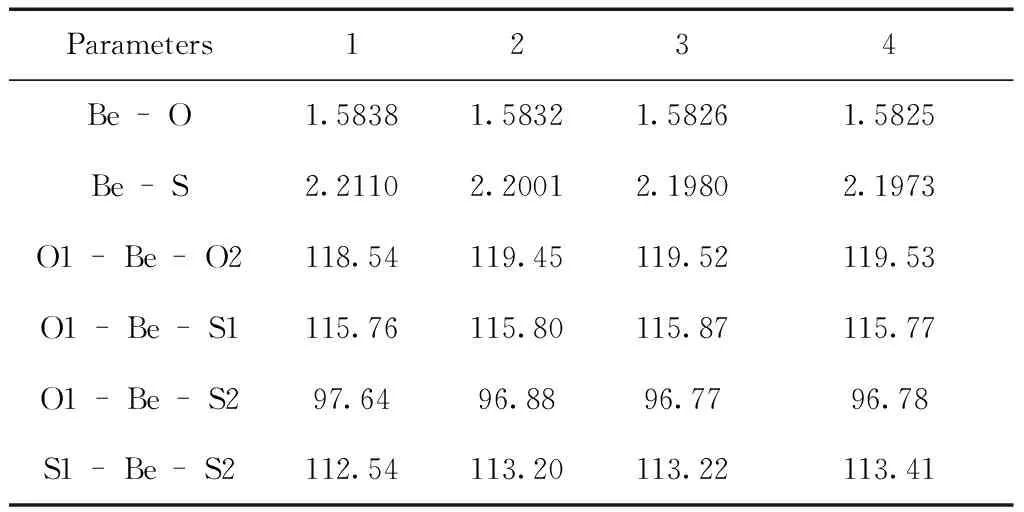
Parameters1234Be-O1.58381.58321.58261.5825Be-S2.21102.20012.19802.1973O1-Be-O2118.54119.45119.52119.53O1-Be-S1115.76115.80115.87115.77O1-Be-S297.6496.8896.7796.78S1-Be-S2112.54113.20113.22113.41
3.2 Be—O/S键的拓扑分析
表2给出了配位键Be—O/S的键临界点处的电子密度拓扑参数. 结果表明4种配合物的Be—O/S的键临界点的ρ(r)均较小,▽2(r)均为正值,说明在键临界点处电荷是发散的,Be—O/S键显示离子键特征,且Be—O键比Be—S键的离子性强.计算的4种配合物的椭圆度ε→0,说明Be—O/S键为σ配键. 另外,Be—S的键临界点处H(r)<0,Be—O的键临界点处H(r)>0,表明Be—S键具有部分的共价性特征,Be—O键共价性明显较低. 与配合物1相比,Be—O/S键强度随烷基给电子能力增大而增大,与键长变化的结果一致.
3.3 前线分子轨道和能级
表3和图2给出了4种配合物分子前线轨道的组成和分布特征. 四个前线轨道中N原子上的H原子和烷基基团以及Be原子的贡献均较少,如Ⅰ%(Be),Ⅳ%(S1)和Ⅴ%(S2)所示. 分子轨道成份主要集中在吡啶环部分和S、O原子上,如Ⅱ%(S1)和Ⅲ%(S2)所示,且均为π轨道特征. 从表3中可以看出,4种配合物的分子轨道能级分布特征极为相似,与配合物1相比,配合物2~4的前线分子轨道能级均有较小幅度的升高,且随烷基给电子能力的增强而增大,但LUMO轨道能级升高幅度比HOMO轨道能级大,结果能隙ΔH-L变大. 配合物HOMO和LUMO能级、能隙大小顺序均为4>3>2>1,与下文TD-B3LYP计算的光学带系的变化趋势一致. 从前线能级分布特征,可以预测配合物1的N原子上的烷基取代物的导电性会降低,其吸收和发射光谱发生蓝移. 由表3和图2所示,4种配合物的HOMO和HOMO-1之间以及LUMO和LUMO+1之间的能级差在0.0315~0.0519 eV之间,其差值较小,表明4种配合物发生HOMO→LUMO电子跃迁时,会伴随HOMO-1→LUMO+1,且均为π→π*跃迁.
表2 配合物1-4各配位键临界点处的电子密度拓扑参数(a.u.)
Table 2 Topological parameters (in a.u.)at bond critical points of complexes 1-4
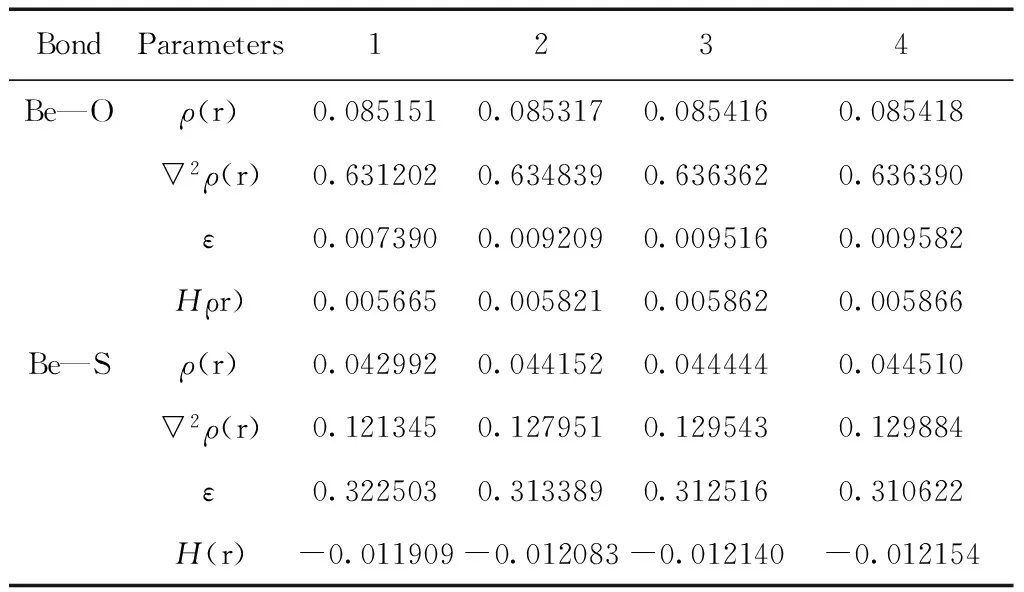
BondParameters1234Be—Oρ(r)0.0851510.0853170.0854160.085418▽2ρ(r)0.6312020.6348390.6363620.636390ε0.0073900.0092090.0095160.009582Hρr)0.0056650.0058210.0058620.005866Be—Sρ(r)0.0429920.0441520.0444440.044510▽2ρ(r)0.1213450.1279510.1295430.129884ε0.3225030.3133890.3125160.310622H(r)-0.011909-0.012083-0.012140-0.012154
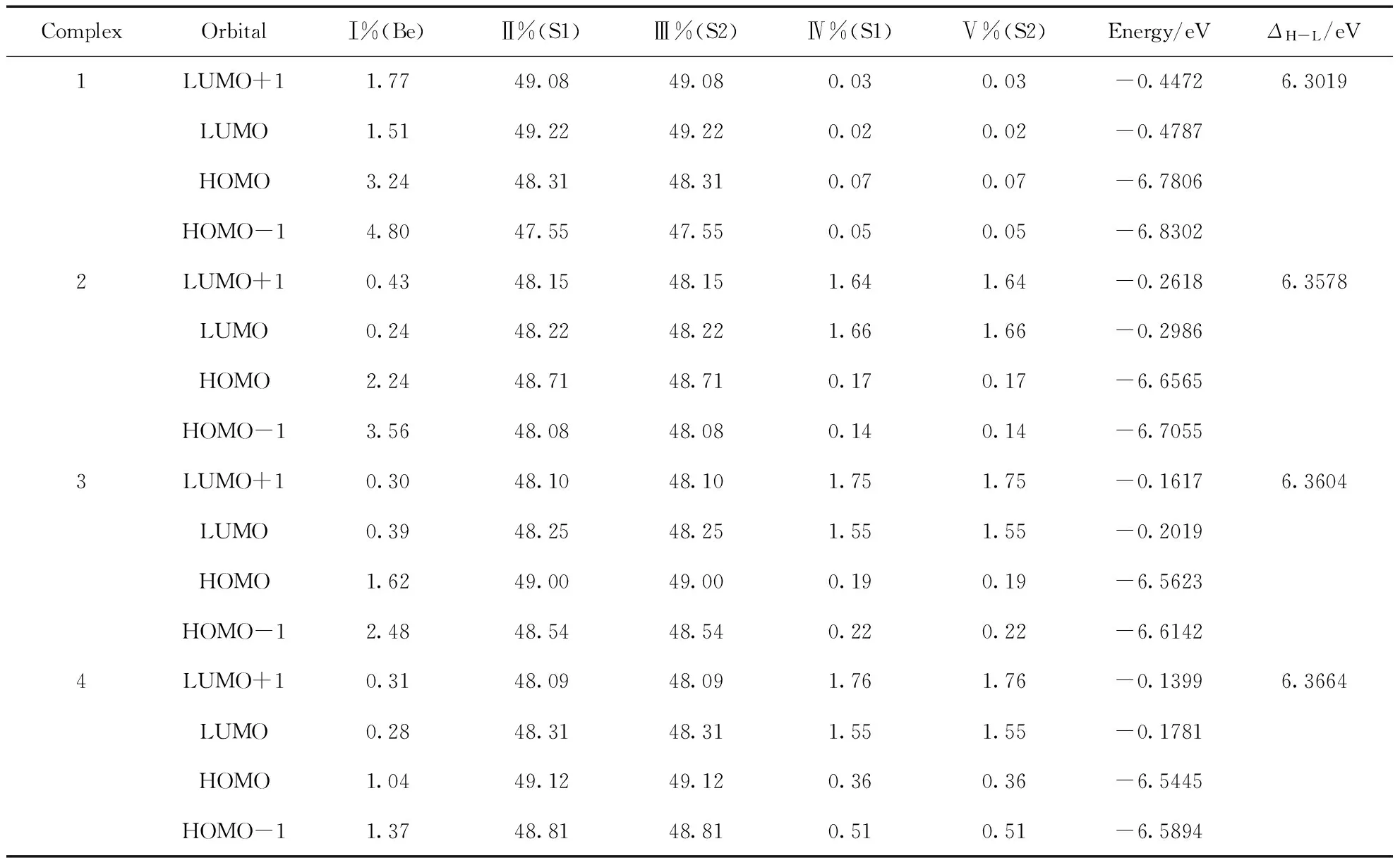
表3 配合物1-4的前线分子轨道的组成和能级及能隙

图2 配合物1-4的前线分子轨道图Fig. 2 The frontier molecular orbitals for complexes 1-4
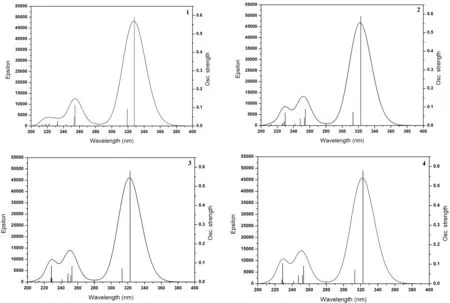
图3 配合物1-4在气相中模拟的吸收光谱图Fig. 3 The simulated absorption spectra of complexes 1-4 in gas media
3.4 电子吸收光谱
在优化的基态几何构型基础上,利用TD-B3LYP方法计算了各配合物的电子吸收光谱.前线轨道分析表明最大吸收波长吸收峰都具有π→π*跃迁特征. 为了更好地理解烷基对配合物的电子结构和光谱性质的影响,计算了4种配合物20个单重激发态. 表4中分别列出了最大吸收波长、振子强度(f)和跃迁组成. 4种配合物的最大吸收波长分别为327.99、322.82、322.72 和322.71 nm,振子强度分别为0.5898、0.5892、0.5801和0.5778,主要是电子从HOMO-1→LUMO+1和HOMO→LUMO的跃迁,且为3-羟基-2-吡啶硫酮配体的本体π→π*跃迁,与前面分子轨道分析讨论的结果一致. 图3为模拟的4种配合物的电子吸收光谱.4种配合物吸收谱图极为相似,在280~380 nm范围内有一分割明显吸收较强的单峰,在200~280 nm有两个相互重叠的吸收较弱两个峰. 与配合物1相比,配合物2~4的最大吸收波长都发生的较小程度的蓝移,这主要是由于4种配合物具有相似的分子构型和电子结构,烷基取代基引入不会对配合物的光谱性能产生明显的影响.
4 结 论
利用DFT和TD-DFT方法研究了4种Be配合物的电子结构和电子吸收光谱的特征. 计算结果表明4种N-取代-3-羟基-2-吡啶硫酮Be配合物中,由于烷基取代基团的修饰,分子的结构、前线轨道及能隙、和电子光谱等都受到了不同程度的影响. 与配合物1相比,其它3种取代物的能隙均增大,吸收光谱发生了蓝移.理论研究结果显示,烷基取代基对该类配合物的结构、性质与功能的影响较小. 本文研究结果可以为设计新型发光金属配合物材料提供理论依据.
表4 配合物1-4的最大吸收波长(λ)、振子强度(f)、及轨道跃迁成份的计算结果
Table 4 The maximum absorption wavelengths (λ), oscillator strengths (f) of complexes 1-4
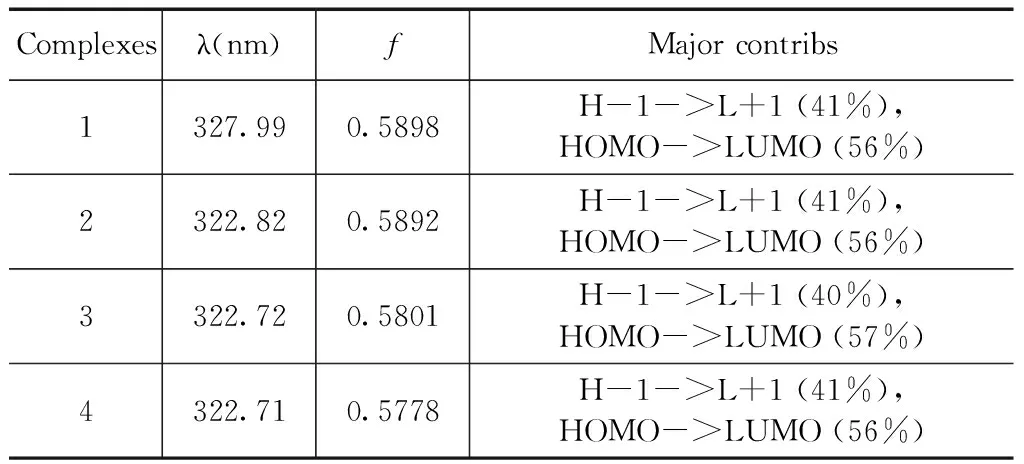
Complexesλ(nm)fMajorcontribs1327.990.5898H-1->L+1(41%),HOMO->LUMO(56%)2322.820.5892H-1->L+1(41%),HOMO->LUMO(56%)3322.720.5801H-1->L+1(40%),HOMO->LUMO(57%)4322.710.5778H-1->L+1(41%),HOMO->LUMO(56%)
[1] Evans R C, Douglas P, Winscom C J. Coordination complexes exhibiting room-temperature phosphorescence: Evaluation of their suitability as triplet emitters in organic light emitting diodes [J].Coord.Chem.Rev., 2006, 250 (15): 2093.
[2] Chi Y, Chou P T. Transition-metal phosphors with cyclometalating ligands: fundamentals and applications [J].Chem.Soc.Rev., 2010, 39 (2): 638.
[3] Wang S. Luminescence and electroluminescence of Al(Ⅲ), B(Ⅲ), Be(Ⅱ) and Zn(Ⅱ) complexes with nitrogen donors [J].Coord.Chem.Rev., 2001, 215 (1): 79.
[4] Halls M D, Schlegel H B. Molecular orbital study of the first excited state of the OLED material tris(8-hydroxyquinoline)aluminum(Ⅲ) [J].Chem.Mater., 2001, 13(8): 2632.
[5] Lewis J A, Cohen S M. Addressing lead toxicity: Complexation of lead(II) with thiopyrone and hydroxypyridinethione O,S mixed chelators [J].Inorg.Chem., 2004, 43(21): 6534.
[6] Katoh A, Yokoyama H, Matsumura Y,etal. Synthesis of metal complexes with 1-substituted 3-hydroxy-2(1H) pyridinethiones and their insulinmimetic activities [J].Heterocycles, 2010, 81(3): 585.
[7] Lewis J A, Tran B L, Puerta D T,etal. Synthesis, structure and spectroscopy of new thiopyrone and hydroxypyridinethione transition-metal complexes [J].DaltonTrans., 2005, 15: 2588.
[8] Frisch M J, Trucks G W, Schlegel H B,etal. Gaussian 09, Rev. D.01. Wallingford, CT: Gaussian, Inc., 2013.
The structures and spectrum properties of Be(Ⅱ) complexes with N-substituted 3-hydroxy-2-pyridinethione ligand
HUANG Rong-Yi, WU Gen-Hua
(Anhui Key Laboratory of Functional Coordination Compounds, School of Chemistry and Chemical Engineering, Anqing Normal University, Anqing 246011, China)
The geometry structures of four selected Be(Ⅱ) complexes were optimized at B3LYP/6-31+G(d) level of density function theory. The effects of the alkyl substituent groups on the geometrical and electronic structures have also been explored. Based on the above stable structure, the electronic absorption spectra for each complex have been theoretically investigated via TD-DFT at the aforementioned basis set level. The computational results indicate that the donation electron ability of alkyl substituent group can change the frontier molecular orbital energy levels, and results in the increases of HOMO-LUMO energy gaps. As a result, the maximum absorption spectra of the three alkyl substituent derivates have a substance blue shift accordingly.
Be(Ⅱ) coordination complex; Electronic structure; Electronic spectra; TD-DFT
103969/j.issn.1000-0364.2015.12.003
2014-10-05
国家自然科学基金(21171008);安徽高校省级优秀青年人才基金(2010SQRL108ZD)
黄荣谊(1975—),男,安徽肥西人,硕士,副教授,主要从事量子化学和功能配合物研究.E-mail: huangry@aqtc.edu.cn
O641.4
A
1000-0364(2015)06-0916-05
猜你喜欢
安徽化工(2022年1期)2022-02-15
石油化工技术与经济(2021年6期)2022-01-18
中华养生保健(2020年3期)2020-11-16
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
中国资源综合利用(2017年1期)2018-01-22
造纸化学品(2017年1期)2017-01-21
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
郑州大学学报(理学版)(2014年3期)2014-03-01
