
缺血再灌注过程中肝细胞自噬研究进展
2014-12-31 09:39:58李静静郭传勇
胃肠病学和肝病学杂志 2014年10期
李静静,郭传勇
同济大学医学院附属第十人民医院消化科,上海200072
自噬(autophagy)是真核细胞利用溶酶体降解损伤的长寿蛋白及细胞器的过程,通过对细胞降解成分的再利用在维持内环境稳态中起着非常重要的作用。正常生理情况下,它维持在较低的水平,清除多余、受损的细胞器,为细胞提供保护作用。在病理情况下,氧化应激、肝细胞营养缺乏、缺血再灌注损伤、肝脏衰竭都能引起自噬,是一种细胞对不利因素的适应性反应。近年来国内外各项研究显示,自噬参与肝脏疾病的多个领域,尤其与缺血再灌注损伤过程关系十分密切,现就近年来关于自噬与肝脏缺血再灌注损伤相关作用和机制的研究进展作一概述。
1 细胞自噬
1.1 概述 自噬现象最早是由Ashford 和Porter[1]于1962 年用电子超微显微镜观察人体肝细胞时发现,是细胞内的一种重要代谢过程。细胞的部分细胞质与细胞器被从粗面内质网的无核糖体附着区脱落的双层膜或多层膜包裹形成自噬体(autophagosome)后与溶酶体整合形成自噬溶酶体,被其分解为核苷酸、氨基酸、游离脂肪酸等小分子供细胞本身的代谢和能量的利用。
1.2 分类 根据生理过程和细胞内底物转运溶酶体方式的不同,可将哺乳动物的自噬现象分为3 种:(1)大自噬(macroautophagy):细胞浆中可溶性蛋白质和坏死的细胞器被非溶酶体来源的双层膜包裹形成自噬小泡,与溶酶体融合后形成氨基酸等被再利用的过程,目前以大自噬的研究为主。(2)小自噬(microautophagy):小自噬直接利用溶酶体包裹吞噬并降解胞浆中的长寿蛋白。(3)分子伴侣介导的自噬(chaperon mediated augophagy,CMA):胞质内蛋白分子的氨基酸序列结合到分子伴侣Hsc73,之后被转运到溶酶体腔中被水解酶分解供细胞再利用。CMA 的底物是可溶的蛋白分子,可选择性清除蛋白质,而其余两种自噬现象无明显的选择性。因此,自噬被认为是真核细胞中存在最广泛的降解-再循环系统。
1.3 分子机制 目前参与自噬的基因有上百种,被统一命名为自噬相关基因(autophagy associated gene,Atg)。一条修饰过程是Atg12 首先被IE 样酶Atg7 活化后转运至Atg10,然后与Atg5 结合形成自噬体前体,最后此复合物与Atg16 结合形成Atg5-Atg12-Atg16L复合物,此过程促进了自噬泡的伸张,使之由小泡样的结构发展为环状结构。同时,Atg5-Atg12-Atg16L 复合物也促进了微管相关蛋白1 轻链3(LC3)向自噬泡的聚集,而LC3 修饰是自噬泡的形成过程的必要组成部分。
第二条为泛素样蛋白加工修饰过程。此过程参与LC3 的形成,LC3 被Atg4 分解生成LC3-Ⅰ,同时LC3-Ⅰ也被E1 样酶Atg7 活化,转运至Atg3,生成自噬体的标志分子-LC3-Ⅱ。如果自噬体与溶酶体融合,自噬体内的LC3-Ⅱ立即被溶酶体中的水解酶降解。哺乳动物细胞中内源性Atg5 和Atg12 主要以结合的形式存在[2],而胞浆可溶性LC3-Ⅰ和膜结合型LC3-Ⅱ之间的比例在不同组织和细胞类型变化巨大。哺乳动物细胞自噬进程中两条泛素样加工的修饰过程可以互相调节,互相影响(见图1)。
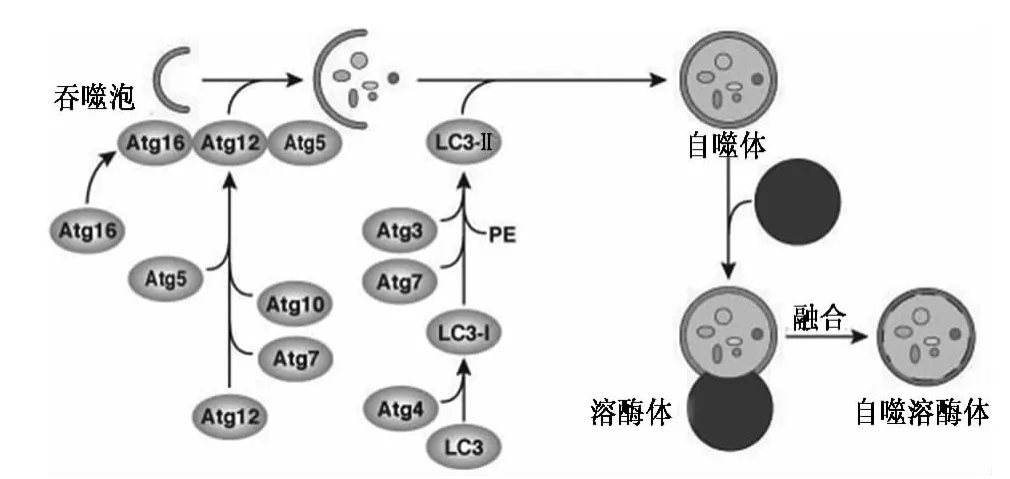
图1 细胞自噬的分子机制Fig 1 The molecular mechanism of autophagy
1.4 发生发展 自噬现象的发生过程以大自噬为例可分为4 个部分:(1)自噬的诱导。在正常条件下,细胞自噬一般在体内以低水平进行用以维持内部环境的平衡稳态,当细胞受到细胞器的损坏、突变蛋白的累聚及微生物的侵袭以及饥饿、营养缺乏等外部应激时,其自噬活性迅速升高。靶向雷帕霉素蛋白(target of rapamycin,TOR)激酶便可使自噬在细胞生长因子存在和营养丰富情况下关闭,感受细胞氨基酸、ATP 和激素的变化,是调控细胞自噬过程的关键点。哺乳动物靶向雷帕霉素蛋白(mammalian target of rapamycin,mTOR)同样以一种类似酵母菌的机制调控自噬过程。除此之外,Ras 信号通路也可通过生长因子调控自噬,可将信号从生长因子酪氨酸激酶传递到细胞内的效应器,如Raf/促分裂素原活化蛋白激酶(mitogen-activated protein kinases,MAPK)和磷脂酰肌醇3 激酶1(phosphatidylinositol 3-kinase 1,PI3K-1)。(2)自噬体的形成。随着分隔膜不断延伸,将被降解的胞浆成分完全隔离形成自噬体。(3)自噬体的运输、融合。自噬体通过细胞骨架微管网络系统将细胞降解成分传送至溶酶体中。(4)内容物的降解利用。随着人们对自噬发生过程研究的深入,新的相关环节不断发现,对医学进步进程的推动起到了巨大作用,但仍有许多重要调控机制及模型有待进一步探究。
1.5 检测方法 目前自噬常用的检测方法大致可分为形态学方法、免疫染色方法以及分子生物学方法。利用电镜观查到膜状结构的自噬小体属于形态学方法,是最主要最直接的证据,是自噬检测的金标准。免疫染色法可将LC3-Ⅰ(微管相关蛋白1 轻链3)经泛素样加工修饰,与磷脂酰乙醇结合形成LC3-Ⅱ[3],通过LC3 组成成分的含量和比例表明自噬发生的数量和程度。近年来,绿色荧光蛋白(GFP)与LC3 结合的质粒已广泛用于研究自噬。
2 肝脏缺血再灌注与自噬
2.1 肝脏缺血再灌注中的自噬现象 通常情况下,机体通过良好的血液循环维持机体的正常代谢及功能。而充血性心力衰竭、休克、肝外伤及心脏手术经常导致心源性及血容量性休克,引起严重低血压和低氧血症进而导致肝脏缺血。在对自噬特异性标志物LC3-Ⅱ的检测中发现,一定条件下的血液再灌注后,部分细胞结构破坏以及功能代谢障碍加重伴随着自噬活性的升高,这对缺血再灌注损伤时肝细胞功能作用的探究具有重大意义。
Sybers 等[4]早在30 年前便首次观察到体外培养的小鼠在类似缺血再灌注的处理后,含有坏死细胞器的自噬泡不断增加。Decker 等[5]也通过兔心模型验证了单纯的缺血现象即可以诱导心肌自噬,而再灌注过程可以强化自噬,同时在另一实验中发现缺血超过1 h 的状态下,无功能性自噬泡数量增加,这为我们进行肝缺血再灌注过程中自噬的研究提供了良好的前提。近期临床实验表明,在人造缺血再灌注条件下,多例患者的治疗效果均有所提高,表明自噬水平的增加可以抑制肝脏细胞的死亡,但自噬对肝缺血再灌注损伤到底起保护还是损伤作用仍存在较大的争议,现在的主要研究都集中在其信号传导与机制方面。如P53、P62、GSK3β、血红素加氧酶1(HO-1)等都对肝脏缺血再灌注中的自噬起到重要作用。
2.2 诱发及调节机制 研究精确的自噬信号转换机制对研究自噬对肝功能恢复的意义至关重要。自噬与其他生物体存在的生理过程一样,都受到细胞以及细胞内成分的调控用以维持机体的稳态平衡,目前作为此机制的中心分子TOR 是控制细胞自噬的关键点,它感受细胞成分与信号的变化,改变自噬的发生发展水平,如能量、酸碱平衡以及PI3K-1、MAPK 及p53 等基因蛋白的水平,都可以通过此途径调节自噬的变化过程(见图2)。
2.2.1 依赖mTOR 途径:TOR 是一个丝氨酸/苏氨酸激酶,调控细胞的周期和生长等。在哺乳动物中,处于活化状态的TOR 同源物mTOR 通过磷酸化抑制自噬过程的起始分子ULK 的功能,从而抑制自噬发生。TOR 可以生成复合体,包括mTOR、GβL(也称为类G蛋白β 亚基蛋白)和Raptor 等,信号传导分子Rash 和PI3K-1 可通过磷酸化Akt(蛋白激酶B)、ERK1/2 和PKC(蛋白激酶C)传递信号到小GTP 酶从而使mTOR活性增强,抑制自噬发生。
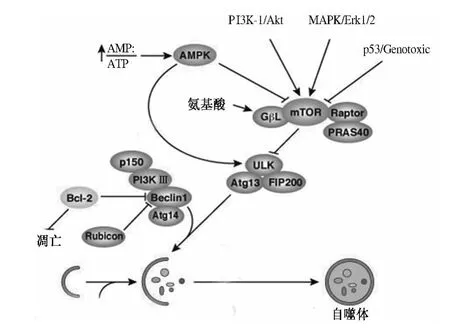
图2 肝细胞自噬的信号传导机制Fig 2 The signal transduction mechanism of autophagy in liver cells
2.2.2 不依赖mTOR 途径:其关键分子是PI3K-Ⅲ/hVps34。同源体Beclin-1 和抑癌基因UVRAG[6]作为自噬的正调控因子,抗凋亡因子Bcl-2 负性调控自噬,共同参与组成复合物调控自噬发生。
2.2.3 AMPK 途径:AMPK 在自噬的发生中也发挥着极其重要的作用,作为细胞中感受能量状态调节代谢的一个蛋白激酶,在低ATP 水平状态下(如饥饿或缺氧),AMPK 感受能量的变化,可通过磷酸化TSC2,抑制mTOR 活性,诱导细胞发生自噬[7]。与此同时也有研究表明,此途径可以直接促进ULK 诱导自噬发生。
2.3 自噬对肝脏缺血再灌注的作用 细胞的程序性死亡对细胞存活具有非常重要的意义,Kohli 等[8]以及国内通过建立大鼠肝脏缺血再灌注模型研究表明,再灌注损伤过程中发生肝细胞凋亡,且随缺血时间延长而加强,从而推断凋亡可促使凋亡分子及其诱导分子释放,使肝脏细胞死亡。但Gujral 等[9]发现通过抑制半胱氨酸天冬氨酸蛋白酶并不能有效地保护肝功能,这就说明另一种细胞死亡机制-自噬在保护过程中发挥重要作用。目前,对心脏缺血再灌注中自噬的功能作用研究已相对比较成熟,但对肝脏还有很大的研究空间,肝细胞缺血与心肌缺血本质上相同,因此,自噬可以被急性和慢性缺血诱导在再灌注过程会促进自噬体的形成。但缺血过程中的自噬对肝脏起保护作用还是损伤作用,目前尚存在较大争议。
LC3 是自噬体的特异性标记蛋白,通过缺血再灌注后LC3 的表达水平可即时反映自噬情况变化,国内研究显示在缺血再灌注30 min 后有自噬体的产生,而在3 h 后达到最高峰,随后在24 h 细胞自噬水平不断降低[10]。结合AST、ALT 对细胞功能的检测,可以认为自噬对肝脏缺血再灌注损伤起某种程度的保护性反应。在肝脏缺血再灌注损伤中,自噬的出现与组织血供情况、刺激强度作用有关,缺血时间短,伤害刺激较弱均可刺激自噬激活,在一定程度上减少细胞凋亡,保护肝功能。而这种关系被Hamacher-Brady 等[11]验证凋亡与自噬是通过线粒体通透性转换(mitochondrial permeability transition,MPT)而实现的。短时间、少量营养物质缺乏导致部分线粒体通透性增加,自噬水平增强,产生负反馈抑制溶酶体降解肝细胞,而大量线粒体发生MPT 时,由于ATP 水解过多,发生大量氧化磷酸化脱耦联,细胞就表现为凋亡、坏死从而引起肝细胞损伤,凋亡与自噬平衡点是众多学者研究的关键。而在此平衡点内采取干预措施控制自噬的水平能减少肝细胞的进一步损伤需要更加深入细致的探究。
体外培养HepG2 细胞[12],通过三气培养箱对细胞进行缺血缺氧再灌注的处理,并在吖啶橙染色荧光显微镜定性观察细胞质内出现红色的点状及斑点状结构为自噬特征性结构-自噬泡,这说明细胞缺血后细胞内自噬水平迅速提高。再灌注恢复其氧供后LC3的变化随时间不同而变化,伴随AST/ALT 酶学检测肝细胞也发生变化。
2.4 自噬相关因子研究进展 目前,国内外多项研究表明,各种酶学及基因对自噬的调节功能各不相同,从酶学角度上说,HO-1(血红素加氧酶1)[13]可通过PI3K 途径提高自噬的发生水平从而减轻肝脏缺血再灌注损伤。而3-甲基腺嘌呤(3-MA)[14]是通过抑制磷脂酰肌醇激酶降低ATP 水平,特异性阻断细胞中自噬发生,干扰肝功能恢复。靶蛋白GSK-3β 被PKB 磷酸化后失活,稳转GSK-3βKD[15]细胞自噬水平高于未转录的细胞。
从基因水平上来说,P53 参与自噬过程的调节,一方面通过活化激活AMPK,负性调节mTOR,另一方面通过上调mTOR 抑制因子或其靶蛋白DRAM 负性调节mTOR,从而诱导发生,但Tasdemir 等[16]通过实验利用P53 抑制剂PTF-α 抑制P53 的活性或用siRNA沉默P53,均可导致细胞自噬作用增强,表明P53 可负性调节自噬,其机制可能与其含量、应激水平或者应激信号有关。最新研究发现P53 参与自噬调节,其中包括转录非依赖途径和转录依赖途径:转录非依赖途径是通过P53 的活化激活AMPK,负性调节mTOR,而转录依赖途径可通过上调mTOR 抑制因子PTEN 和基因TSC1,或是P53 调节的自噬和细胞死亡基因DRAM 负性调节mTOR,从而诱导自噬发生。
3 总结与展望
自噬与疾病关系的研究已经成为此领域一个新的热点。目前,对于自噬在肝脏缺血再灌注过程中发挥的作用还没有明确的结论,在正常肝缺血的早期,自噬可能发挥对细胞的保护作用,应以此为依据,通过加强药物干预和效果监测调动自噬在恢复肝细胞功能中的保护作用,相反在疾病发展的中晚期,由于其与细胞凋亡的串扰[17]以及负反馈调节的失衡,自噬对肝细胞起损伤作用。在不同疾病中,自噬也发挥相区别的作用[18],肝癌中自噬诱导细胞程序性死亡而抑制其保护作用。
因此,提高肝缺血再灌注治疗中自噬的水平,探寻其发展、调节因素和调节通路,掌握自噬与其凋亡等生理过程的相互关系,有助于我们发现新的治疗肝病的策略。例如,作为组织中对缺血、缺氧最敏感的内皮细胞在自噬条件下生理状态如何变化,P53 的双层作用以及与调控有关的各因子的影响因素,对于这些机制更加深入的认识,终将为人类对抗和最终解决这些疾病问题产生深远影响。
[1] Ashford TP,Porter KR. Cytoplasmic components in hepatic cell lysosomes[J]. J Cell Biol,1962,12:198-202.
[2] Tanida I,Ueno T,Kominami E. LC3 conjugation system in mammalian autophagy [J]. Int J Biochem Cell Biol,2004,36 (12):2503-2518.
[3] Chen Y,Azad MB,Gibson SB.Methodsfor detecting autophagy and determining autophagy-induced cell death[J]. Can J Physiol Pharmacol,2010,88(3):285-295.
[4] Sybers HD,Ingwall J,DeLuca M. Autophagy in cardiac myocytes[J].Recent Adv Stud Cardiac Struct Metab,1976,12:453-463.
[5] Decker RS,Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes[J]. Am J Pathol,1980,98(2):425-444.
[6] Maejima Y,Kyoi S,Zhai P,et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2[J]. Nat Med,2013,19(11):1478-1488.
[7] Jung CH,Ro SH,Cao J,et al. mTOR regulation of autophagy[J].FEBS Lett,2010,584(7):1287-1295.
[8] Kohli V,Selzner M,Madden JF,et al. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver[J]. Transplantation,1999,67(8):1099-1105.
[9] Gujral JS,Farhood A,Jaeschke H. Oncotic necrosis and caspase-dependent apoptosis during galactosamine-induced liver injury in rats[J]. Toxicol Appl Pharmacol,2003,190(1):37-46.
[10] Zhang Y. The role of cell apoptosis and autophagy in rat liver ischemia-reperfusion injury[D]. Fudan University,2008.张勇. 细胞凋亡与自体吞噬在大鼠肝脏缺血再灌注损伤中的作用研究[D]. 复旦大学,2008.
[11] Hamacher-Brady A,Brady NR,Logue SE,et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy [J]. Cell Death Differ,2007,14(1):146-157.
[12] Ye L,Zhao X,Lu J,et al. Knockdown of TIGAR by RNA interference induces apoptosis and autophagy in HepG2 hepatocellular carcinoma cells[J]. Biochem Bilphys Res Commun,2013,437(2):300-306.
[13] Morse D,Lin L,Choi AM,et al. Heme oxygenase-1,a critical arbitrator of cell death pathways in lung injury and disease [J]. Free Radic Biol Med,2009,47(1):1-12.
[14] Boya P,González-Polo RA,Casares N,et al. Inhibition of macroautophagy triggers apoptosis [J]. Mol Cell Biol,2005,25 (3):1025-1040.
[15] Wang SH,Shih YL,Kuo TC,et al. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells[J]. Toxicol Sci,2009,108(1):124-131.
[16] Tasdemir E,Maiuri MC,Orhon I,et al. p53 represses autophagy in a cell cycle-dependent fashion[J]. Cell Cycle,2008,7 (19):3006-3011.
[17] Gordy C,He YW. The crosstalk between autophagy and apoptosis:where does this lead?[J]. Protein Cell,2012,3(1):17-27.
[18] Shi F,Wang MR. The research progress of autophagy and its relationsip with tumors [J]. Chinese Journal of Cell Biology,2011,33(12):1366-1373.石峰,王明荣. 细胞自噬及其与肿瘤关系的研究进展[J]. 中国细胞生物学学报,2011,33(12):1366-1373.
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07 06:21:30
昆明医科大学学报(2022年2期)2022-03-29 00:52:18
汽车维修与保养(2021年8期)2021-02-16 00:28:20
生物化工(2021年2期)2021-01-19 21:28:13
生物化工(2020年1期)2020-02-17 17:17:58
读与写(2019年35期)2019-11-05 09:40:46
现代职业教育·高职高专(2018年7期)2018-05-14 16:20:40
工业设计(2016年4期)2016-05-04 04:00:15
癌变·畸变·突变(2016年3期)2016-02-27 06:15:36
哈尔滨医药(2015年4期)2015-12-01 03:57:54
