
富勒烯与β淀粉样肽低聚体结合的分子动力学模拟
2014-10-18 05:28周晓颖郗文辉韦广红
物理化学学报 2014年8期
周晓颖 郗文辉 韦广红
(应用表面物理国家重点实验室,计算物质科学教育部重点实验室,复旦大学物理系,上海 200433)
1 引言
一系列重大疾病与蛋白或多肽的淀粉样聚集有关,如阿兹海默氏症(AD)、帕金森症、二型糖尿病等.1,2其中阿兹海默氏症,由于患者众多,一直备受人们的关注.3,4AD患者会出现出一系列神经退行性症状,如记忆功能退化、语言及认知能力的丧失等,严重时将致死.AD发病几率随着年龄增长呈现几何级数增长,在美国AD已经成为仅次于心脏病和癌症的第三大致死疾病.由于目前尚没有治疗AD的特效药物,对AD的研究受到各界人士的广泛重视.5,6通常认为AD的成因主要与大脑内的Aβ、Tau等蛋白的错误折叠和淀粉样聚集密切相关.Aβ是一种通过多种分泌酶剪切淀粉质前体蛋白而得到的多肽,长度约为37-43个氨基酸,其中以Aβ40和Aβ42这两种具有神经细胞毒性的淀粉样聚集体最为常见.7-9Aβ40在大脑内的浓度数倍于Aβ42,但后者具有更强的毒性.正常状态下Aβ并不具有特定的三维结构,其本身的生理功能也尚不清楚;然而在病理状态下,Aβ会在大脑内形成淀粉样纤维并大量沉淀,最终形成光学显微镜下可见的斑块.10通过电镜、固相核磁共振(ss-NMR)等多种实验手段,人们已经大致了解该淀粉样聚集体的分子结构特征.11-13Aβ40/42通过链间平行β片,排列成可以无限延伸的原纤维;进而多条原纤维之间互相缠绕,最终形成电镜下可观察到的细长纤维状的聚集体.14有实验表明,Aβ在聚集过程中形成的低聚物和纤维均具有神经毒性,15是导致AD的原因之一.研究如何破坏或抑制Aβ的淀粉样聚集,对理解Aβ的聚集机理以及开发治疗AD的方案具有重要意义.
虽然Aβ可以在试管中自发从可溶单体状态聚集为淀粉样沉淀,但是在细胞内,Aβ处于一个复杂的溶液环境,将受到诸多因素的影响,如pH、16,17小分子18或纳米颗粒19-21等.不同因素会对Aβ聚集产生促进或抑制作用,因此若干治疗策略都是基于某种调控机制来抑制或破坏Aβ的聚集过程.22碳纳米颗粒,作为纳米颗粒的一种,具有如工艺成熟、表面修饰便利、生物毒性低等一系列优点,在药物运输、表面催化等领域均有广泛运用.23-25不少富勒烯或石墨烯等碳纳米颗粒相关的实验和模拟侧重于淀粉样聚集蛋白体系.26-29最近一系列的研究工作表明,碳纳米颗粒的曲率等物理化学性质将对淀粉样多肽及非淀粉样多肽的吸附和聚集产生一定的影响.27,30,312003年Kim和Lee32发现修饰后的富勒烯可以有效抑制全长Aβ的聚集,这一结果为开发C60类抗聚集药物提供了良好的指引方向.Andujar等33利用分子对接研究了富勒烯与Aβ的结合位点,得到了在有序的β-sheet顶部的位点为能量有利的结合位点,并采用分子动力学模拟研究了富勒烯在此结合位点上与Aβ的相互作用及其对Aβ结构的破坏,此结果为理解富勒烯的抑制效应提供了一些初步的思路,但有一些不清楚的地方,例如:(1)如果不用对接方法,而是采用分子动力学模拟,从富勒烯与Aβ没有接触的状态出发,是否能找到更多的结合位点?(2)从此状态出发富勒烯结合Aβ的动力学过程是怎样的?(3)富勒烯在不同位点上对Aβ低聚体的结构有什么样的影响?这些问题均有待进一步的深入研究.
为了回答上述问题,本工作中利用分子动力学(MD)模拟的方法,从富勒烯与Aβ没有接触(即二者之间的原子间的最近距离为2.0 nm)的状态出发,研究了富勒烯与Aβ42聚集体的结合及相互作用的动力学过程.我们不仅得到了与之前研究结果吻合的位点,而且发现了新的重要位点.在核心疏水区域,观察到C60挤入多肽表面疏水口袋的现象.在N端的位点中,富勒烯具有破坏其末端β结构的能力.通过分析结合的动力学特征及结合能,发现了富勒烯在淀粉样多肽表面结合所特有的沟槽滚动机制.这些结果对理解富勒烯抑制Aβ聚集的聚集机制给出了更全面的解释,为相关抗聚集药物分子的设计提供了有益的思路.
2 模拟方法
2.1 Aβ42与富勒烯的模拟体系

图1 Aβ42六聚体与富勒烯模拟体系的三种初始结构Fig.1 Three different initial conformations of Aβ42 hexamer-fullerene system
本文中的分子模拟选取Aβ42和富勒烯C60分子作为研究体系,见图1.其中多肽体系选取1-42全长Aβ的六聚体,其序列见图1(d).Aβ的聚集体具有多种形貌,实验上通过各种手段已经获得若干种Aβ的聚集体结构.所有结构均具有U turn形状且链间平行β片结构的共性,但细节却存在差异.13如N端部分的结构,有些工作认为N端的若干残基处于无规构象,12,34而另一些工作则解出N端具有结构特征,35,36这种多结构与淀粉样多肽聚集形貌的多样性是吻合的.本文中采用了Ma和Nussinov36于2006年提出的Aβ1-42结构,该结构的特征是Aβ的N端的1-16号残基形成β片结构,而17-42号残基的结构则与Luhrs等12的结果相似.我们选取了该结构的六聚体作为研究对象.相对于富勒烯分子,多肽体系的尺寸可以近似认为是无限长的原纤维.C60的结构和参数则使用Roccatano等37于2011年得到的结果.
为了消除初始构象对模拟采样的影响,我们选取了三种不同的初始状态,见图1中(a-c).三种初始构象中,多肽的初始结构完全相同,C60与多肽体系的最近距离均为2 nm,但放在了不同的位置.在初始构象(c)中,C60放置于Aβ的生长轴方向,而(a,b)中则位于与(c)方向垂直的平面内.水盒子的边界设为2 nm,由于模拟采取了周期边界条件,C60位于相反方向的初始构象也可以被考虑在内.从每个初始构象出发,进行了10条模拟,共计30条.作为对照组,没有C60的纯多肽体系也单独进行了相同条件的模拟.
2.2 分子动力学模拟细节及分析方法
本工作使用全原子层次的分子动力学模拟(MD)及显式水模型.所有的模拟均采用Gromacs 4.5.3软件包38计算,多肽体系选用GROMOS 43a1力场39参数,溶液分子则使用简单点电荷(SPC)模型.40静电计算使用了Particle-Mesh-Ewald(PME)方法,41静电及范德华作用的截断半径分别为0.9和1.4 nm,因此初始构象中多肽和C60之间没有直接相互作用.蛋白体系及溶液分子的键长分别使用LINCS算法42及SETTLE算法43加以约束,故积分步长设为2 fs.整个模拟过程中,温度始终保持310 K,压强为一个标准大气压.由于不同的模拟中多肽与C60结合的过程不同,因此模拟的时间也各不相同.所有的模拟都在二者紧密结合不再分开后至少延长5 ns,故模拟时间从10 ns到40 ns不等,详情见后,而对照组的多肽体系则模拟了50 ns.
所有的轨迹均采用VMD软件仔细加以观察监测.所有的分析均采用AmberTool2013工具包44内提供的工具完成,结合位点的分析统一采用每条轨迹中C60结合多肽后的最后5 ns轨迹.二级结构倾向性(SSP)分析采用DSSP算法;45而分子的溶剂可及表面积(SASA)使用Connolly46提出的方法.Cluster构象分析则使用Averagelinkeage算法,47对应的代表结构均为cluster分析方法得到的代表构象(Representative structure).在所有的分析中,水分子均已剔除.
为了计算富勒烯的结合能,我们运用了分子力学-泊松波尔兹曼表面积(MM-PBSA)方法,并使用广义Born模型(GB model)方法计算溶剂化效应.48MM-PBSA计算方法见下述公式(1).体系的结合能ΔG等于复合物自由能Gcomplex减去对应单体的自由能Gligand和Greceptor,故结合能包括(见下面公式(2)):真空中的范德华相互作用能ΔEvdW和静电能ΔEcoul、极性和非极性溶剂化自由能(ΔGGB,ΔGsurf)以及振动熵ΔS的贡献.在模拟中,由于富勒烯上的碳原子不带电,故第二项静电能为零;第三项通过GB方法计算得到,而第三项与第四项之和则为总的溶剂化效应.Amber的MM-PBSA方法中计算振动熵的算法并不适用于本工作的体系,44且以前类似的工作中也略去这一项的贡献,33故本文中不予考虑.所有力场参数均被转化为Amber格式以保持一致,并利用idecomp方法计算了每个残基对结合能的贡献.49

3 结果与讨论
3.1 结合的动力学过程
在所有的模拟中,不论是否存在C60,Aβ的主链β结构均没有发生严重破坏,绝大多数主链间氢键在模拟的过程中保持稳定.在所有的模拟中,富勒烯经过不同的时间,最终均能够到达多肽表面,并且与其紧密结合.在我们的工作中,当富勒烯与多肽重原子之间的最近距离小于0.4 nm(略大于两个碳原子的vdW半径之和)并持续1 ns以上时,认为富勒烯已结合在多肽上.图2展示了不同轨迹中二者结合所需的时间.为了便于对比,此图中将不同轨迹按照结合所需时间的长短进行了排序.可以看出不同的模拟中,结合所需的时间差别很大,从约1 ns到大于30 ns不等.图2中三种不同线型的曲线分别表示不同的初始构象,可以看出结合的时间与系统的初始构象没有关系.这从一个方面说明我们的模拟采样较好.
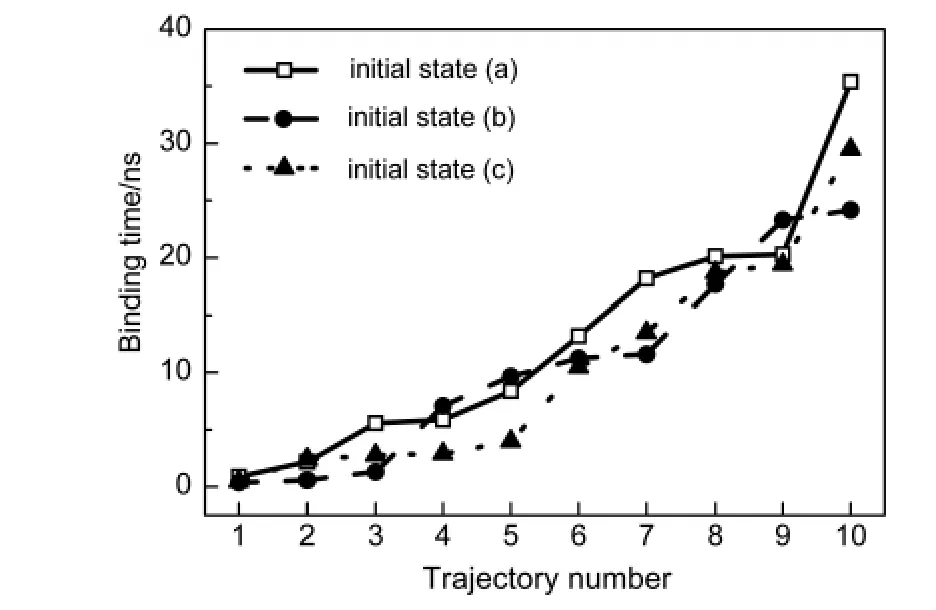
图2 不同模拟轨迹中C60结合到Aβ多肽上所用的时间Fig.2 Time for fullerene binding to Aβ hexamer in different simulated trajectories
通过观察所有的模拟轨迹,发现C60在靠近多肽的初期,会在其表面发生小幅度的滚动,有时还可以离开多肽.经过若干次的尝试过程,最终C60将到达某个位置与蛋白紧密结合.一旦到达这种结合状态,富勒烯的运动幅度将大大降低,通常只能在附近0.5 nm的幅度内摇摆,不会再离开多肽表面.图3展示了两条不同的模拟轨迹中系统的构象变化(a,b)及对应轨迹中C60与多肽最近原子之间的距离.我们分别计算了多肽与富勒烯复合物体系(实线)和多肽(虚线)的均方根偏差(RMSD),而参考结构为最后5 ns模拟的cluster聚类代表结构.可以看出多肽的结构一直处于平均约0.3 nm的涨落之中;整个体系的均方根偏差在开始时变动幅度较大,而在后半段中则与代表多肽的红线几乎重合,这说明结合前C60在不断移动,而结合后则稳定在其结合位点附近,只发生很小幅度的涨落.而从图3(c,d)中可以看出,在最终结合前,富勒烯分子可以不止一次地靠近多肽,并且再次离开.这说明C60在蛋白表面的结合是具有选择性的,不够好的结合位置将被抛弃.在所有的模拟中,这种紧密结合状态至少可以稳定存在5 ns.我们从每个初始构象随机选择一条轨迹,在其结合到多肽之后,继续延长30 ns的模拟,结果显示在这个时间尺度内C60仍然不会离开多肽表面.因此我们认为达到了这种稳定结合态后,系统的状态将趋于平衡,结合位点不会再发生大幅度变化,将该结合位置定义为C60结合的一个位点.这说明一定时间长度的模拟即可表征这种结合态,下文中针对结合态的分析均基于稳定结合后的最后5 ns数据.
3.2 结合位点的分类及结合能
本工作的关注点之一为采用分子动力学模拟的方法研究富勒烯与Aβ的结合位点,此外,两者的结合机制也是研究的重点.将所有30条模拟的结合态中C60每个时刻的质心位置计算出来,一起显示在多肽表面,结果见图4.图中多肽结构选取的是对照组中50 ns模拟的cluster中心结构,而每个C60的质心则用一个紫色点显示.可以看出结合位点具有下述特征.首先,结合位点并非随机分布在多肽表面,而是集中在某几类区域;其它位置则很少出现或完全没有.如Aβ的核心疏水区(CHC)17-21号残基、βturn位置附近(22-30号残基)、C端的纤维生长方向上等.这意味30个结合位点可以归为有限的几类.我们根据C60结合位置附近的氨基酸序号,将其分为如下6类位点:N-terminal 1-5位点、Y10-H14位点、CHC位点(16KLVFFA21)、Turn 24-26位点、Turn 27-31位点、C-terminal位点等.另有2条轨迹中的结合位置只出现了一次,分别位于F4H6附近及N端7号氨基酸附近,故不予归类而统称others.所有类型的结合位点均在图4中用箭头标明.此处CHC位点与前人得到的结果是一致的,而由于对方使用的Aβ结构中N端1-16号残基并非β结构的缘故,其N端区域的位点与我们的结果没有可对照性.33

图3 Aβ-fullerene体系的均方根偏差及Aβ和C60最小距离(d min)随时间演化Fig.3 Time evolution of RMSD ofAβ-fullerene complex and the minimum distance between Aβ and C60(d min)
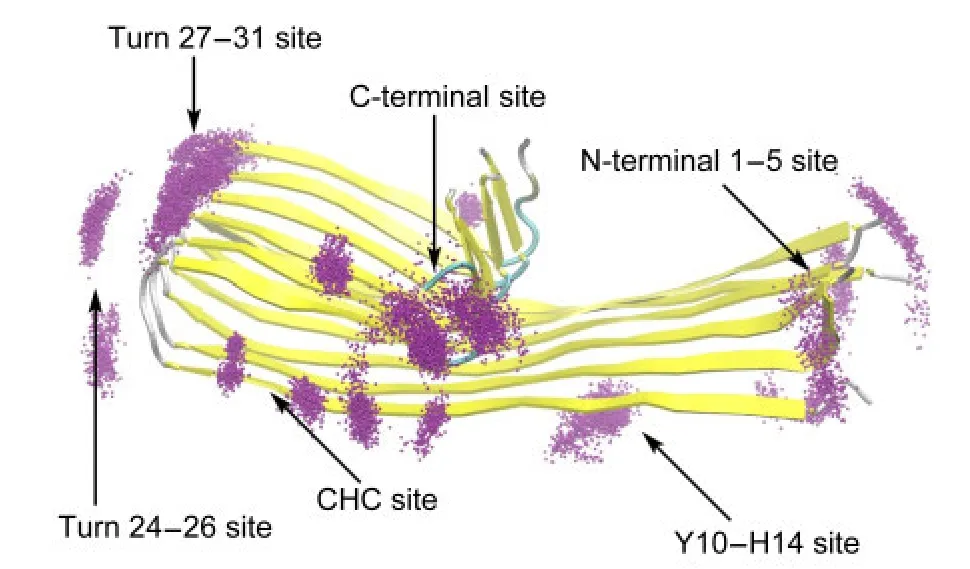
图4 所有的结合位点上C60的质心位置分布Fig.4 Positions of fullerene′s mass center at all binding sites
其次,结合位点的分布表明了结合位点的振动范围.在多数模拟轨迹中,C60的摆动幅度极小,如CHC位点及C-terminal位点;而N-terminal位点看似摆动幅度较大,实则是多肽末端自身的振动所导致的,故该位点实际的摆动幅度与CHC位点类似;有趣的是Turn端两个位点及Y10-H14位点,富勒烯的晃动幅度明显高于其它位点.C60的摆动方向沿着纤维的生长方向,由于淀粉样聚集体结构中的重复性,当其摆动时,C60在肽链间的相同位置平移,这种平移运动并不需要克服大的能量势垒.
另外,通过观察所有轨迹,发现除一条轨迹外,几乎不存在结合后结合位置跨位点的移动.在仅有的一个例外中,观察到富勒烯从C-terminal位点缓慢转移到了CHC位点,并稳定于此.考虑到C-terminal位点与CHC位点在空间位置上距离较近,这一结果并不意外.没有出现大规模的跨位点转移行为一方面说明了位点分类的合理性,另一方面也暗示各个结合位点之间存在一定的能量势垒,C60想要从一个结合的好的位置通过表面滚动到达另一个较好的位点,要跨越的能量代价较大,故极少观察到这种现象.
为了进一步理解各个结合位点之间的差异,同时分析各种相互作用在结合机制中所起的作用,我们计算了所有位点的结合能,结果见表1.在所有的位点中,范德华作用对于稳定结合起主导作用,而因为设定所有C60上的碳原子电荷为零,故二者间没有直接静电作用项.溶剂的效应由GB作用(ΔGGB)和表面能(ΔGsurf)共同构成,总值大于零,这与富勒烯的强疏水性是吻合的.从总的结合能可以看出结合最强的两个位点为Turn 27-31和CHC位点;其它类的两个位点的结合能也较强.N-terminal、Y10-H14及C-terminal这三个位点的结合能则很接近,Turn 24-26位点最弱.另外结合能的均方差较小,这表明每个位点分类中的各个轨迹之间的结合能相近,从另一个方面再次论证了位点分类的合理性.在前人的工作中,33CHC位点是结合能最强的位点,这与我们的结论是基本吻合的,而数值上的不同则归因于所使用的力场之间的巨大差异.
为了进一步验证模拟不受初始构象的影响,我们将三种初始构象所得到各个结合位点的数量列出,结果见图5,可以看出从不同初始构象出发能得到同一个位点,从同一个初始构象出发可得到多类位点,说明结合位点对初始构象的依赖性不大.这进一步说明了模拟采样的结果较好.
3.3 富勒烯对Aβ42核心疏水区间结构的影响
Kim和Lee32的实验工作中观测到了C60对Aβ聚集有抑制效果.因此我们的目的并非仅限于分析结合位点,而是期望能够理解其抑制聚集的机制.他人的模拟工作中,限于分子对接方法的局限性,并不能全面回答这一问题.33在我们的模拟工作中虽然没有看到C60大规模破坏Aβ结构的现象,然而,在CHC位点的一条轨迹中,我们观察到了富勒烯挤入CHC位置附近疏水口袋的趋势.为了更加详细地研究这个现象,我们将CHC位点的五条模拟轨迹在C60结合后延长了20 ns.如图6(a-d)所示,当碳球结合到CHC位点后,碳球逐渐挤入两层β片之间,与位于从F19到I32的整个β-turn区域的氨基酸均有或多或少的接触,尤其是侧链方向朝向内部的F19、A21、K28、A30及I32,并降低这一带残基二级结构的稳定性.通过分析碳球的溶剂可及表面积,可以发现C60在~1.5 ns时结合到该位点,之后富勒烯通过不断调整位置,挤入多肽CHC区域,期间伴随着其SASA的变化,最后溶剂可及表面积稳定在约1.2 nm2,如图6(e)中的红线所示.可见富勒烯确实有挤入Aβ结构内部的趋势.从图6(c,d)中可以看出,在20和30 ns时C60对多肽turn端结构的破坏作用大于10 ns时的,turn处的几个残基偏离附近的多肽而靠向富勒烯分子.并非所有CHC位点均存在这种现象,通过计算另外四条CHC位点轨迹中C60的SASA(图6(e)中的蓝、黑、灰、绿线),可以看出其它模拟中C60的SASA在结合后保持稳定,并没有显著降低趋势.
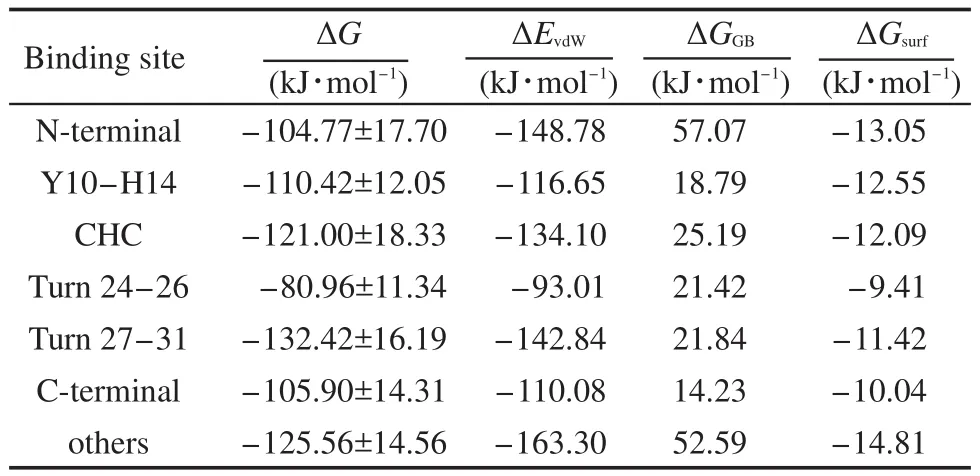
表1 富勒烯在每个位点的结合能Table 1 Binding energy of fullerene at each binding site

图5 从不同初始构象出发得到的各结合位点轨迹数目Fig.5 Counts of trajectories leading to different binding sites,starting from different initial states
这一钻孔现象为我们提供了一种抑制聚集机制的可能解释:即由于CHC位点上氨基酸的疏水性,多肽能与C60之间产生极佳的结合性.当C60结合到该位点时,挤入该疏水口袋会提供更好的疏水匹配,减少疏水表面积,从而提高溶剂分子的构象熵.且二者之间的结合很紧密,因此不会大幅度降低侧链间的疏水堆积效应.考虑到CHC区域对Aβ聚集的关键作用,50C60挤入这一位置将会对原纤维的生长产生阻碍效应.

图6 C60挤入Aβ原纤维核心疏水区域的趋势Fig.6 Tendency for C60to insert into the CHC region of Aβ protofibril

图7 C60对Aβ蛋白N端β片结构的破坏Fig.7 Disruption of the β-sheet structure of Aβ peptides′N-terminal region by C60
3.4 富勒烯对蛋白N端二级结构的破坏
在所有三十条模拟中,没有观察到大幅度的Aβ二级结构破坏.然而在N-terminal位点的某条轨迹中,发现了富勒烯对Aβ结构的局部破坏现象,其结果见图7.其中图7(a-d)展示了不同时刻系统的构象,在t=21 ns时,C60开始结合到该位点,此时2-6号的氨基酸仍然保持其β二级结构;随着时间演进,C60将Aβ最外侧的链逐渐拉开,一个个的破坏其链间的主链氢键,从而破坏其β结构,从t=29 ns时刻可以看到这种破坏作用已经开始发生,对氢键有了一定程度的破坏.为了进一步观察这种破坏效应,将该模拟延长了30 ns,结果发现这个时间尺度内破坏效应没有进一步增加的趋势.图7(e)中展示了最外侧Aβ1-10号残基的β片百分比随时间的变化,在尚未有直接作用前,3-10号残基均以β片构象为主,而富勒烯与多肽结合后,3-5号残基的β片依次随时间降低,最终完全破坏;而7-9号残基的β结构亦略有下降.而在30-60 ns时间段,β结构没有显著的变化.
这一β结构破坏过程完整地展示了一种富勒烯破坏Aβ聚集体结构的机制:首先结合至Aβ聚集体生长面的N端残基,进而与N端残基的链间氢键竞争,逐步破坏氢键,拉开β片;破坏几个氢键后,就挤入两条肽链之间,阻止氢键的复原.由于Aβ自身结构的稳定性和短模拟时间的局限性,模拟尚不能采样到破坏整条Aβ的二级结构的事件,这可能与模拟中所采用的富勒烯的低浓度有关,在实验中,当C60的浓度很低时,其抑制聚集的效果亦不显著.可以推想随着富勒烯浓度的增高,多个C60会同时结合到这一位点,必将大大增强对主链氢键的破坏作用,同时多个C60的结合可以更有效地阻止β结构的复原.这一抑制机制很可能与上节中CHC位点的机制相辅相成,从不同路径破坏Aβ自身的结构稳定性,从而发挥其抑制聚集的效应.
3.5 富勒烯在淀粉样纤维表面的沟槽滚动机制
由于淀粉样纤维中结构单元的重复性,聚集蛋白或多肽能够形成具有周期性的表面形貌.当某些位置的氨基酸主链及侧链处于一种特定的构象时,将形成沿着生长轴方向的沟槽,其大小恰好可以容纳富勒烯分子.我们在Aβ42聚集体表面的Turn 27-31位点和Y10-H14位点观察到了这一沟槽,见图8(a,c).在Turn 27-31位点N27、K28及G29的主链形成一个很好的弯曲,配合N27指向外侧的侧链,恰好产生一个能够容纳C60的弧形区域.由于G29、A30都是没有侧链或侧链很小的残基,且甘氨酸具有很强的主链构象柔性,故这一位置主链的柔软和侧链的缺失为形成半径合适的沟槽提供了很好的帮助.而在Y10-H14位点,则通过指向两侧的Y10和V12的侧链形成这一沟槽,H14的侧链则从旁辅助.Y10与V12均具有很强的疏水性,且Y10的侧链芳香环能够与富勒烯直接接触,通过芳香环堆积作用形成较强的相互作用,故这一位点的沟槽更多的是靠侧链的疏水作用及特异相互作用实现的.
通过观察模拟轨迹,发现在这两个位点的模拟中,C60的晃动幅度比其它位点要大,可以在同一氨基酸位置从链二到链五来回摆动,从图4位点分布中可以证实这一点,在这两个位点大量紫色的点沿着纤维生长方向排布在多肽表面,形成狭长的带状分布,而与其它位点的质心分布形式大不相同.为了进一步分析这种沟槽内的滚动现象,我们分析了每个残基所有位点的平均结合能,见图8(e),其中红色的区域表示结合能强,蓝色则反之.可以看出在Y10-H14及N27-I31残基位置形成了独特的纵向带状条纹.为了便于理解,图8(b)和(d)中将对应位点附近每个残基按照其平均结合能着色,对应氨基酸的侧链已标明.一个有趣的现象是在这两个位点中,处于中部的肽链对应残基的结合能强于两侧末端肽链,这表明位于这一沟槽内的富勒烯滚动时,将不倾向于到达末端而是偏好于停留在中部.末端这一能量势垒将阻止C60从纤维的中部跨越到生长面上,从而隔离开这两个位点与生长面上的位点,如CHC和C-terminal位点.

图8 C60结合到Aβ原纤维表面的沟槽部位的倾向Fig.8 Tendency of C60binding to the groove on the surface of Aβ protofibril
这种沿着蛋白表面沟槽滚动的现象是淀粉样聚集蛋白所独有的特征;因为只有周期重复性的结构,才能形成可延伸的重复侧链排布,从而形成这种沟槽.C60作为典型的完全对称的球状小分子,在周期性侧链形成的表面沟槽内滚动时,几乎不存在能量势垒,从而能够很轻易地移动.考虑到所有的淀粉样聚集蛋白均能在表面形成或多或少的各种半径的沟槽,这一特殊的“沟槽滚动机制”具有普遍意义,并在预测其它聚集多肽与碳球类衍生物的聚集行为及结合位点时提供一定的帮助.
4 结论
本工作采用分子动力学模拟系统地研究了富勒烯在Aβ42聚集体表面的结合过程,并得到了多类结合位点.C60与Aβ的结合并不是随机的,而是在经历一系列尝试过程后,最终找到某个稳定的结合位点.绝大多数情况下结合后C60将稳定在结合位点附近,而某几类位点中富勒烯能够沿着纤维生长轴方向大幅度滚动.共得到六类主要结合位点,其中CHC位点及Turn 27-31位点具有最强的结合稳定性.二者之间的结合通过范德瓦尔斯相互作用稳定,而溶剂化效应则起相反作用.
在两个位点中观察到了C60对Aβ二级结构的破坏.其一位于核心疏水区域,C60具有挤入多肽两片β片中间的趋势,从而降低附近残基的β结构稳定性,这一位点不但在前人的结果中也发现过,并且早已被认为对聚集过程具有重要作用;其二位于N端末尾,C60能够破坏纤维生长端Aβ的3-5号残基的主链氢键,从而破坏其β片结构,这一直接破坏过程对于理解富勒烯抑制Aβ聚集机制提供了很好的解释.实际过程中两种机制很可能相辅相成,共同发挥抑制聚集的作用.
另外,发现了富勒烯与淀粉样多肽聚集体结合的一种特殊机制,即沟槽滚动机制.由于淀粉样聚集体的结构重复性,蛋白表面沿纤维生长方向能形成特定形貌的沟槽,而富勒烯因其结构的对称性,能够在槽内滚动.这一机制有助于预测富勒烯在其它淀粉样肽聚集体表面的结合位点及其结合行为.
(1)Chiti,F.;Dobson,C.M.Annu.Rev.Biochem.2006,75,333.doi:10.1146/annurev.biochem.75.101304.123901
(3)Selkoe,D.J.Physiol.Rev.2001,81,741.
(4)Selkoe,D.J.Jama-J.Am.Med.Assoc.2000,283,1615.doi:10.1001/jama.283.12.1615
(5)Blennow,K.;de Leon,M.J.;Zetterberg,H.Lancet 2006,368,387.doi:10.1016/S0140-6736(06)69113-7
(6)Selkoe,D.J.Nature 1999,399,A23.
(7)Hardy,J.;Selkoe,D.J.Science 2002,297,353.doi:10.1126/science.1072994
(8)LaFerla,F.M.;Green,K.N.;Oddo,S.Nat.Rev.Neurosci.2007,8,499.doi:10.1038/nrn2168
(9)Reinhard,C.;Hebert,S.S.;De Strooper,B.Embo.J.2005,24,3996.doi:10.1038/sj.emboj.7600860
(10)Finder,V.H.;Glockshuber,R.Neurodegener.Dis.2007,4,13.doi:10.1159/000100355
(11)Rauk,A.Chem.Soc.Rev.2009,38,2698.doi:10.1039/b807980n
(12)Luhrs,T.;Ritter,C.;Adrian,M.;Riek-Loher,D.;Bohrmann,B.;Doeli,H.;Schubert,D.;Riek,R.Proc.Natl.Acad.Sci.U.S.A.2005,102,17342.doi:10.1073/pnas.0506723102
(13)Tycko,R.Annu.Rev.Phys.Chem.2011,62,279.doi:10.1146/annurev-physchem-032210-103539
(14)Straub,J.E.;Thirumalai,D.Annu.Rev.Phys.Chem.2011,62,437.doi:10.1146/annurev-physchem-032210-103526
(15)Walsh,D.M.;Selkoe,D.J.J.Neurochem.2007,101,1172.doi:10.1111/j.1471-4159.2006.04426.x
(16)Ma,K.;Clancy,E.L.;Zhang,Y.B.;Ray,D.G.;Wollenberg,K.;Zagorski,M.G.J.Am.Chem.Soc.1999,121,8698.doi:10.1021/ja990864o
(17)Peralvarez-Marin,A.;Barth,A.;Graslund,A.J.Mol.Biol.2008,379,589.doi:10.1016/j.jmb.2008.04.014
(18)Liu,F.F.;Dong,X.Y.;Sun,Y.Acta.Phys.-Chim.Sin.2010,26,1643.[刘夫锋,董晓燕,孙 彦.物理化学学报,2010,26,1643.]doi:10.3866/PKU.WHXB20100613
(19)Saraiva,A.M.;Cardoso,I.;Pereira,M.C.;Coelho,M.A.;Saraiva,M.J.;Mohwald,H.;Brezesinski,G.ChemBioChem 2010,11,1905.doi:10.1002/cbic.201000237
(20)Bitan,G.;Kirkitadze,M.D.;Lomakin,A.;Vollers,S.S.;Benedek,G.B.;Teplow,D.B.Proc.Natl.Acad.Sci.U.S.A.2003,100,330.doi:10.1073/pnas.222681699
(21)Cabaleiro-Lago,C.;Quinlan-Pluck,F.;Lynch,I.;Dawson,K.A.;Linse,S.ACS Chem.Neurosci.2010,1,279.doi:10.1021/cn900027u
(22)Cummings,J.L.New Engl.J.Med.2004,351,56.doi:10.1056/NEJMra040223
(23)Kolosnjaj,J.;Szwarc,H.;Moussa,F.Toxicity Studies of Fullerenes and Derivatives.In Bio-Applications of Nanoparticles;Springer:New York,2007;p 168.
(24)Geckeler,K.E.;Samal,S.Polym.Int.1999,48,743.doi:10.1002/(SICI)1097-0126(199909)48:9<743::AID-PI246>3.0.CO;2-4
(25)Jensen,A.W.;Wilson,S.R.;Schuster,D.I.Bioorgan.Med.Chem.1996,4,767.doi:10.1016/0968-0896(96)00081-8
(26)Li,C.X.;Mezzenga,R.Nanoscale 2013,5,6207.doi:10.1039/c3nr01644g
(27)Todorova,N.;Makarucha,A.J.;Hine,N.D.M.;Mostofi,A.A.;Yarovsky,I.PLoS Comput.Biol.2013,9(12),e1003360.
(28)Podolski,I.Y.;Podlubnaya,Z.A.;Kosenko,E.A.;Mugantseva,E.A.;Makarova,E.G.;Marsagishvili,L.G.;Shpagina,M.D.;Kaminsky,Y.G.;Andrievsky,G.V.;Klochkov,V.K.J.Nanosci.Nanotechnol.2007,7,1479.doi:10.1166/jnn.2007.330
(29)Huang,H.M.;Ou,H.C.;Hsieh,S.J.;Chiang,L.Y.Life Sciences 2000,66,1525.doi:10.1016/S0024-3205(00)00470-7
(30)Guo,J.;Li,J.;Zhang,Y.;Jin,X.;Liu,H.;Yao,X.Plos One 2013,8,e65579.
(31)Zuo,G.;Zhou,X.;Huang,Q.;Fang,H.;Zhou,R.J.Phys.Chem.C 2011,115,23323-23328.doi:10.1021/jp208967t
(32)Kim,J.E.;Lee,M.Biochem.Biophys.Res.Commun.2003,303,576.doi:10.1016/S0006-291X(03)00393-0
(33)Andujar,S.A.;Lugli,F.;Hofinger,S.;Enriz,R.D.;Zerbetto,F.Phys.Chem.Chem.Phys.2012,14,8599.doi:10.1039/c2cp40680b
(34)Petkova,A.T.;Yau,W.M.;Tycko,R.Biochemistry 2006,45,498.doi:10.1021/bi051952q
(35)Lu,J.X.;Qiang,W.;Yau,W.M.;Schwieters,C.D.;Meredith,S.C.;Tycko,R.Cell 2013,154,1257.doi:10.1016/j.cell.2013.08.035
(36)Ma,B.Y.;Nussinov,R.Curr.Opin.Chem.Biol.2006,10,445.doi:10.1016/j.cbpa.2006.08.018
(37)Hezaveh,S.;Samanta,S.;Milano,G.;Roccatano,D.J.Chem.Phys.2011,135(16),164501.doi:10.1063/1.3643417
(38)Hess,B.;Kutzner,C.;van der Spoel,D.;Lindahl,E.J.Chem.Theory Comput.2008,4,435.
(39)van Gunsteren,W.F.;Billeter,S.;Eising,A.;Hünenberger,P.H.;Krüger,P.;Mark,A.E.;Scott,W.;Tironi,I.G.Biomolecular Simulation:the GROMOS96 Manual and User Guide;Vdf Hochschulverlag AG an der ETH Zürich:Zürich,1996.
(40)Berendsen,H.;Postma,J.;Van Gunsteren,W.;Hermans,J.Intermolecular Forces 1981,11,331.
(41)Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.1993,98,10089.doi:10.1063/1.464397
(42)Hess,B.;Bekker,H.;Berendsen,H.J.;Fraaije,J.G.J.Comput.Chem.1997,18,1463.doi:10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
(43)Miyamoto,S.;Kollman,P.A.J.Comput.Chem.1992,13,952.doi:10.1002/jcc.540130805
(44)Case,D.A.;Darden,T.A.;Cheatham,T.E.,III;Simmerling,C.L.;Wang,J.;Duke,R.E.;Luo,R.;Walker,R.C.;Zhang,W.;Merz,K.M.;Roberts,B.P.;Wang,B.;Hayik,S.;Roitberg,A.;Seabra,G.;Kolossvai,I.;Wong,K.F.;Paesani,F.;Vanicek,J.;Liu,J.;Wu,X.;Brozell,S.R.;Steinbrecher,T.;Gohlke,H.;Cai,Q.;Ye,X.;Wang,J.;Hsieh,M.J.;Cui,G.;Roe,D.R.;Mathews,D.H.;Seetin,M.G.;Sagui,C.;Babin,V.;Luchko,T.;Gusarov,S.;Kovalenko,A.;Kollman,P.A.AMBER 11;University of California:San Francisco,2010.
(45)Kabsch,W.;Sander,C.Biopolymers 1983,22,2577.doi:10.1002/bip.360221211
(46)Connolly,M.L.J.Appl.Crystallorg.1983,16,548.doi:10.1107/S0021889883010985
(47)Shao,J.Y.;Tanner,S.W.;Thompson,N.;Cheatham,T.E.J.Chem.Theory Comput.2007,3,2312.
(48)Onufriev,A.;Bashford,D.;Case,D.A.J.Phys.Chem.B 2000,104,3712.
(49)Kopitz,H.;Zivkovic,A.;Engels,J.W.;Gohlke,H.ChemBioChem 2008,9,2619.doi:10.1002/cbic.200800461
(50)Tjernberg,L.O.;Näslund,J.;Lindqvist,F.;Johansson,J.;Karlström,A.R.;Thyberg,J.;Terenius,L.;Nordstedt,C.J.Biol.Chem.1996,271,8545.doi:10.1074/jbc.271.15.8545
猜你喜欢
大学物理(2022年9期)2022-09-28
物理通报(2020年7期)2020-07-01
唐山师范学院学报(2020年6期)2020-04-16
中国化妆品(2019年4期)2019-11-20
橡塑技术与装备(2018年10期)2018-05-18
温州大学学报(自然科学版)(2016年1期)2016-10-27
厦门大学学报(自然科学版)(2016年4期)2016-08-04
原子与分子物理学报(2015年3期)2015-11-24
物理化学学报(2015年5期)2015-02-28
应用化工(2014年7期)2014-08-09
