
不同载体对负载型氧化钨催化剂在己二酸合成反应中的结构及性能影响
2014-10-18 05:28:26张召艳祝全敬戴维林宗保宁
物理化学学报 2014年8期
张召艳 祝全敬 丁 靖 戴维林,* 宗保宁
(1复旦大学化学系,上海市分子催化和功能材料重点实验室,上海 200433;2中国石化石油化工科学研究院,催化材料与反应工程国家重点实验室,北京 100083)
1 引言
以WO3为活性组分的多相催化剂,由于具有酸性和氧化还原性能而广泛应用在烯烃的选择氧化、1-4烷烃的异构化、5氮氧化合物的选择还原6等反应中.此外,作为一种光催化剂,WO3还有很多潜在的应用,例如能量转换、7,8病毒失活、9有机污染物降解10,11等,由于其能带间隙较窄(2.7-2.8 eV),在可见光下就可以表现出催化活性.12-14众所周知,这些负载型催化剂中WO3的分散度和存在形式是影响其催化性能的主要因素.在催化剂的制备过程中,不同的制备方法、不同的制备条件、不同的载体可以得到WO3分散度、酸性和存在形态不同的催化剂,从而导致催化剂结构和性能上的差异.
迄今为止,已有很多文献报道了WO3在不同载体上的分散情况以及存在状态.15-19Kim等20研究了WO3在商业SiO2上的存在状态和分散情况,通过浸渍法合成的W-Si催化剂,其表征结果说明以不同的钨源为前驱体时,WO3在SiO2上的存在状态也不相同.以钨酸铵为前驱体时,在W-Si催化剂中出现了晶态WO3;而以W(η3-C3H5)4为前驱体时,没有晶态WO3出现,室温下钨物种以[W12O42]12-多聚钨物种形态存在.Zhu等21通过浸渍方法制备了WO3/ZrO2催化剂,探讨了催化剂中钨物种的存在状态和性质对邻苯二甲醇选择氧化制备苯酞反应中催化性能的影响,发现当钨物种主要以高度分散或无定形状态存在,并且含有多聚[WO6]单元时,催化剂表现出很好的催化活性,提高焙烧温度会导致晶态WO3的出现,使得催化活性降低.Mallesham等22通过对合成的W掺杂SnO2催化剂的研究,发现W物种可以成功地掺杂到SnO2晶格中,进而引起SnO2的晶格收缩,生成了大量的缺陷位和酸性位,提高了催化剂在甘油缩醛反应中的催化活性.Klepel等23则在碱性条件下合成了W-MCM-41介孔分子筛.他们认为,碱性条件可以有效地防止钨物种在W-MCM-41介孔分子筛骨架外形成多聚态钨物种和晶态WO3.紫外-可见漫反射表征表明进入分子筛骨架中的钨物种主要以孤立或低聚态的形式存在.Koo等24成功合成了负载WO3纳米颗粒的MCM-48催化剂,此催化剂对于烯烃、含硫有机物和环酮等的氧化反应具有很高的活性和选择性.然而,该类介孔硅分子材料负载的钨基催化剂仍然存在以下缺点:钨物种的分散度难以控制,甚至负载量较低时,也出现细小的晶态三氧化钨;在高温焙烧或者氧化钨负载量较高时,材料的介孔结构容易出现堵塞或坍塌;而焙烧温度过低会导致钨物种不能完全活化.因此选择合适的载体,对于钨物种的存在状态和催化剂的结构形式至关重要.此外,在双氧水体系中,钨物种的溶脱是普遍存在的问题.因此,探索不同载体与活性物种的相互作用,使得催化剂可以多次重复利用也是研究的重点.牛新书等25采用化学共沉淀法合成了SnO2/WO3粉体,他们发现SnO2可以抑制WO3晶粒的生长,提高了其气体灵敏度.Kamata等26报道了W/Zn/SnO2多相催化剂在烯烃及胺类等的选择氧化反应中有非常高的活性和选择性,并且催化剂可以多次重复使用.由此可见,WO3和SnO2之间存在较强的相互作用.近年来,WO3/SnO2复合材料作为催化剂也开始受到广泛的关注.
己二酸(AA)是一种重要的有机化工原料,主要应用于塑料、树脂、食品等领域,可用于制造尼龙-6,6、增塑剂、润滑脂、聚氨酯泡沫塑料,少量用作食品的增酸剂和代替酒石酸用于发酵粉,也可作为中间体用于制造杀虫剂和黏合剂,以及医药、香料等的生产.27然而,传统己二酸的生产工艺主要是硝酸氧化法,该方法使用强氧化性的硝酸作为氧化剂,生产过程设备腐蚀严重,并产生大量的N2O污染物.以过氧化氢水溶液氧化环己烯合成己二酸的副产物只有水,可以实现清洁生产,具有良好的应用前景.但是,目前报道的催化剂主要是均相钨酸催化剂,催化体系中大多使用有机溶剂、相转移剂、助剂或有机酸配体,而这些添加剂使得反应不符合绿色工艺要求,同时产物的分离、提纯和催化剂再生变得困难.28-30也有报道以硅材料或其他材料为载体,负载钨催化剂进行己二酸的合成反应,但是这些工艺仍存在转化率、选择性不高或催化剂重复利用不好等问题.31-33
本文制备了一系列不同载体的负载型氧化钨催化剂,研究了载体对于钨物种的存在状态的影响.首次报道了不同载体负载型氧化钨催化剂在环氧环己烷选择氧化制备己二酸反应中的催化性能,同时获得了一种高活性、高选择性和高稳定性的催化剂,为己二酸的绿色工艺开发提供了一条新颖的方法.
2 实验部分
2.1 试剂与仪器
2.1.1 试 剂
黄钨酸(WO3·H2O,分析纯),草酸(C2H2O4·2H2O,分析纯),草酸亚锡(SnC2O4,化学纯),正硅酸乙酯(TEOS,分析纯),无水乙醇(C2H5OH,分析纯),过氧化氢(50%水溶液,分析纯),浓盐酸(37%,分析纯),十二胺(DDA,化学纯),P123(EO20PO70EO20,Aldrich,平均分子量5800),氢氧化钠(NaOH,分析纯).
2.1.2 仪 器
透射电镜/场发射透射电镜(TEM/FETEM)采用仪器型号为JEOL JEM 2011型,工作电压为200 kV.X射线衍射(XRD)采用德国Bruker公司的D8 Advance型X射线粉末衍射仪进行催化剂样品的物相分析,射线源采用波长为0.15405 nm的Cu Kα射线,采用Goebel镜将发散X光束汇聚为平行光,管电压为40 kV,管电流为40 mA,扫描速率10(°)·min-1.X射线光电子能谱(XPS)样品测试采用Perkin-Elmer PHI 5000C ESCA System X射线光电子能谱仪,以Mg Kα射线(1253.6 eV)作为光源,分析器通能为93.9 eV,分析室的压力<10-7Pa,X射线靶电压为14.0 kV,功率为250 W.样品压片后测试,所有结合能均以污染碳(C 1s,284.6 eV)进行校正.采用SHIMADZU UV-2450型紫外-可见分光光度计进行紫外-可见漫反射光谱(UV-Vis DRS)实验.将粉末样品装入样品池中,以BaSO4为参比测定,分辨率0.1 nm.扫描范围200-800 nm,扫描速率40 nm·min-1.激光拉曼测试在法国Jobin Yvon型共焦显微激光拉曼光谱仪上进行,分辨率小于3 cm-1,激光能量为12.5 mW(样品表面为2.5-3.8 mW),采用He-Ne激光器,激光波长为632.8 nm.傅里叶变换红外(FTIR)光谱分析采用美国Nicolet公司的Avatar-360红外光谱仪,KBr压片,扫描范围4000-400 cm-1,分辨率为0.9 cm-1,扫描次数32次.
2.2 催化剂的制备
SnO2:将草酸亚锡在923 K空气中焙烧3 h(升温速率为2 K·min-1)得到SnO2.
WO3/SnO2:采用浸渍法负载WO3.将0.5388 g H2WO4和5.4378 g H2C2O4·2H2O加入到250 mL烧杯中,然后加入215 mL去离子水,363 K下搅拌溶解,然后将2 g焙烧得到的SnO2载体分散到此溶液中,363 K下搅拌5 h,然后蒸干水分.所得固体在373 K下烘干过夜,然后在空气气氛的马弗炉中以2 K·min-1的速率升温至923 K焙烧3 h.所得粉末即为20%(质量分数,w)WO3/SnO2.
六角介孔二氧化硅(HMS)分子筛:DDA/EtOH/H2O/TEOS以摩尔比0.27:9.09:29.6:1.0投料,室温下搅拌该混合物直至溶液澄清.在剧烈搅拌下滴加TEOS得到白色胶状混合物.滴加完毕后保持搅拌15 min,室温静置老化18 h.然后过滤,用蒸馏水和乙醇洗涤后在373 K烘箱中烘干.在923 K温度下空气中焙烧5 h去除DDA模板剂,制得HMS分子筛.
WO3/HMS:与WO3/SnO2的负载方法一样,仅把载体换为HMS.所得粉末即为20%WO3/HMS.
介孔分子筛(SBA-15):于313 K下,在60 mL浓HCl和312 mL水的盐酸溶液中加入12 g P123模板剂,搅拌4 h,再加入25.6 g TEOS,搅拌24 h,然后移至水热釜中368 K下晶化3天,取出,过滤、洗涤、干燥,于空气中缓慢加热到773 K,并维持6 h,得到SBA-15分子筛.
WO3/SBA-15:与WO3/SnO2的负载方法一样,仅把载体换为SBA-15.所得粉末即为20%(w)WO3/SBA-15.
2.3 催化活性测试
在25 mL圆底烧瓶中加入0.29 g负载型钨基催化剂,然后加入33 mmol的50%(w)双氧水溶液,室温搅拌,最后加入10 mmol环氧环己烷,升温至338 K,反应2.5 h后升温至363 K继续反应,反应后产物通过酸碱中和滴定的方法进行定量分析.分离得率的测定采用如下方法进行.采用将反应混合物热过滤的方法除去催化剂,在冰箱中冷却反应混合物过夜,抽滤得到己二酸晶体,用冰水洗涤晶体二次,然后烘干后称重,并测定熔点.催化剂重复使用方法是将反应后的催化剂离心分离,依次用去离子水和乙醇洗涤三次后在烘箱中373 K干燥3 h后备用.
3 结果与讨论
3.1 催化剂的结构表征
3.1.1 XRD分析
图1给出了纯SnO2,WO3块体及不同载体制得的负载型氧化钨催化剂的XRD图谱.从图中可以看出,负载后的催化剂中都出现了归属于单斜WO3的(002)、(020)和(200)晶面的衍射峰,但是不同载体负载的WO3的结晶衍射峰强度不同.众所周知,XRD衍射峰的宽度反映了样品的结晶程度.22WO3/SnO2催化剂中 WO3的(002)、(020)和(200)晶面的衍射峰强度最弱,半峰宽较宽,说明WO3的结晶程度很低,晶粒尺寸较小.牛新书等25研究SnO2-WO3粉体时发现SnO2可以抑制WO3的晶粒生长,说明SnO2和W物种之间存在强烈的相互作用,载体SnO2可以抑制WO3的晶化和团聚.此外,在WO3/SnO2的XRD图谱中出现了归属于四方晶系金红石结构的SnO2的(110)、(101)和(211)晶面的衍射峰(JCPDS No.41-1445).当以HMS为载体时,WO3/HMS催化剂中WO3的晶态衍射峰相对较强,而WO3/SBA-15中晶态WO3衍射峰强度最强,半峰宽最小,说明负载在SBA-15上的WO3结晶程度最高,并且晶粒尺寸最大,导致其催化性能降低,与活性结果一致.
3.1.2 TEM和FETEM分析

图1 SnO2、WO3及WO3负载在不同载体上所得催化剂的XRD图Fig.1 XRD patterns of SnO2,WO3,and the supported tungsten oxides prepared from different supports

图2 不同载体负载催化剂的TEM(a,b,c)及FETEM(d)照片Fig.2 TEM(a,b,c)and FETEM(d)images of catalysts with different supports
为了考察活性组分在载体表面的分散状态,我们对不同载体的催化剂进行透射电镜表征,结果见图2.从图2中可以看出,在HMS载体表面,WO3/HMS催化剂中出现了晶态WO3,并且晶粒尺寸较大(约100 nm左右);而在SBA-15表面,可以发现大量晶态WO3的聚集,说明钨物种在SBA-15表面发生团聚,分散性很差.图2(c)显示了以SnO2为载体时WO3/SnO2催化剂的电镜图,从图中不能明显看见WO3物种,但是通过场发射透射电镜图(图2(d))可以明显地看见WO3物种高度分散在SnO2载体表面,并且颗粒尺寸只有2 nm左右.相对于全硅介孔分子筛HMS(1160 m2·g-1)和 SBA-15(734 m2·g-1)来说,SnO2的比表面积(13.4 m2·g-1)很小,但是W物种却能很好地分散在其表面而没有发生团聚,说明SnO2与W物种之间具有强烈的相互作用,是分散氧化钨物种的良好载体.负载氧化钨物种后,比表面积均有所降低,但氧化硅系列催化剂其比表面积还是远远大于WO3/SnO2催化剂.程序升温还原分析发现,由于SnO2的可还原性,无法准确利用该技术研究钨物种与载体的相互作用.
3.1.3 UV-Vis DRS分析
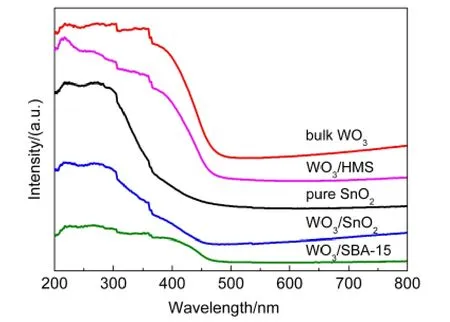
图3 SnO2、WO3及不同载体负载催化剂的紫外-可见漫反射光谱Fig.3 UV-Vis diffuse reflectance spectra of SnO2,WO3,and catalysts with different supports
紫外-可见漫反射技术是研究过渡金属元素离子配位情况的有效表征手段,34通过紫外-可见漫反射表征,可以更好地说明三种催化剂中钨物种的存在状态及分散情况.图3显示了不同载体催化剂的紫外-可见漫反射谱图,其中给出了晶态WO3和SnO2载体的紫外-可见漫反射光谱作为参照.对于全硅介孔分子筛SBA-15和HMS,紫外可见漫反射图谱中没有检测到明显的特征谱峰,当负载WO3后,在230、250-350和450 nm处均出现了吸收峰,分别对应于孤立的类似于单聚钨氧四面体[WO4]的钨物种、低聚态的钨物种以及晶态WO3的特征峰.35-38从WO3/HMS和WO3/SBA-15催化剂紫外可见漫反射图谱上可以看见,在450 nm处有明显的晶态WO3吸收峰,说明钨物种主要以晶态WO3的形式存在,这与XRD和TEM结果相一致.但对于WO3/HMS催化剂来说,在230和250-350 nm处的吸收峰强度比WO3/SBA-15催化剂的要强,说明在WO3/HMS催化剂中单聚的钨氧四面体[WO4]物种和低聚的钨物种的含量要比WO3/SBA-15催化剂多,这些钨物种主要是催化剂在焙烧过程中,钨物种通过与载体相互作用而形成的.20对于WO3/SnO2催化剂,在230 nm和250-350 nm处也观察到了吸收峰,说明WO3/SnO2催化剂中也存在单聚的钨氧四面体[WO4]物种和低聚的钨物种,此外与SnO2载体的紫外可见漫反射谱相比,在WO3/SnO2中并没有明显观察到晶态WO3的吸收峰,说明以SnO2为载体时,催化剂中不存在明显的晶态WO3,钨物种高度分散在SnO2载体上,这一结果与FETEM观测到的现象一致.另外,我们发现催化剂中SnO2的吸收峰向长波方向移动(与纯SnO2相比),这可能是钨物种与载体SnO2间强烈的相互作用所致.
3.1.4 Raman光谱分析
拉曼测试是一种非常灵敏的检测表面氧化钨颗粒的技术,尤其是对于晶态的氧化钨非常有效.因此,Raman光谱无疑是用于检测细小晶态WO3粒子的最有效的手段.如图4所示,晶态WO3表现出四个特征拉曼峰,分别位于804、714、327和267 cm-1处.对于全硅分子筛HMS和SBA-15,由于自身荧光效应很强,因此其拉曼信号很弱,未出现特征拉曼峰.39对于SnO2来说,其拉曼谱峰也很弱,只在634 cm-1处可以看见归属于金红石型SnO2的微弱拉曼峰.40虽然三种不同载体的负载型催化剂都表现出了晶态WO3的拉曼峰,但是峰的位置与晶态WO3拉曼峰相比都发生了一定的位移,可能是WO3物种与载体间相互作用的结果.另外,以SnO2为载体的WO3/SnO2催化剂与WO3/HMS和WO3/SBA-15催化剂相比,WO3的拉曼峰强度最弱,半峰宽最宽,说明在WO3/SnO2中WO3的结晶性能最弱,SnO2可以有效地抑制WO3的晶粒生长;而WO3/SBA-15的拉曼峰强度最强,半峰宽最窄,说明其结晶程度最高,SBA-15载体不利于WO3的分散,导致WO3的聚集,这一发现与XRD和TEM所得结果相一致.
3.1.5 XPS分析
表1列出了不同载体负载氧化钨催化剂的表面钨锡(硅)原子摩尔比和投料比.从表中可以看出,WO3/SnO2催化剂中表面W/Sn摩尔比要远高于投料比,说明钨物种主要存在于SnO2载体表面并且高度分散,而当以HMS和SBA-15为载体时,催化剂表面W/Si摩尔比反而远小于其投料比,说明钨物种在载体表面团聚或者进入孔道,表面分散性不好,并且,SBA-15为载体时分散性最差,这与前面的电镜和XRD结果相一致,说明SnO2是分散钨物种的良好载体.
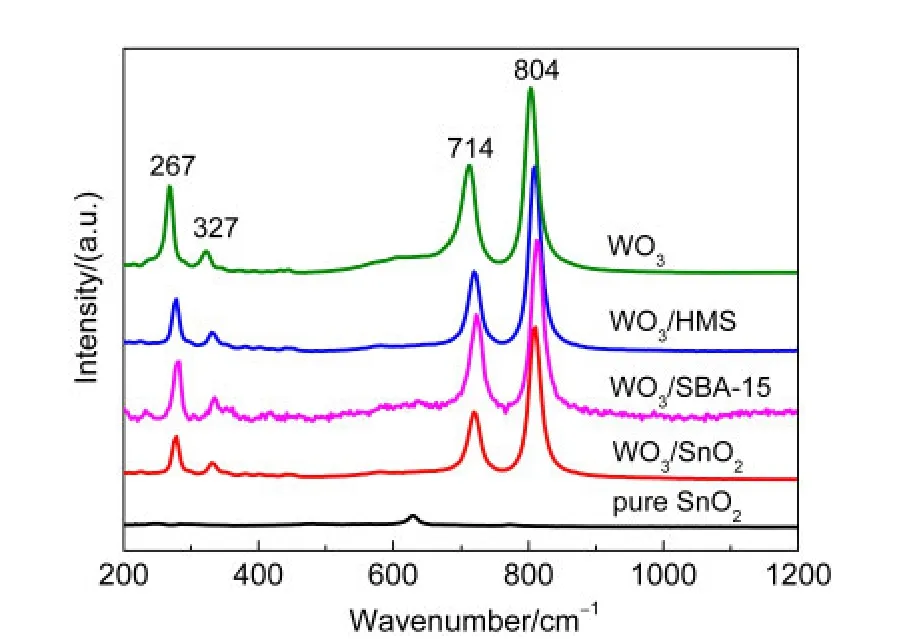
图4 SnO2、WO3及不同载体负载催化剂的拉曼光谱Fig.4 Raman spectra of SnO2,WO3,and catalysts with different supports

表1 不同载体负载催化剂的表面原子W/载体摩尔比Table 1 Surface W/support molar ratio of catalysts with different supports
3.1.6 FTIR分析
在合成含杂原子的负载型催化剂的研究中,红外表征是检测杂原子是否进入载体骨架中的一个有力证据.图5给出WO3和不同载体催化剂的FTIR图谱.对于全硅分子筛作为载体的催化剂中,在460、810、960及1100 cm-1处出现了硅氧振动有关的红外吸收峰,其中460、810及1100 cm-1处的红外谱峰分别对应于硅氧四面体骨架中的Si―O―Si的弯曲振动、对称伸缩振动和反对称伸缩吸收.39另外结合WO3的红外谱图可知,在767和825 cm-1附近出现了W―O的振动峰,因此在全硅分子筛作为载体的催化剂中,W―O的振动峰被Si―O―Si的振动所掩盖.对于960 cm-1的归属却颇有争议,有的文献将其归属于Si―OH的红外振动峰,41但是也有研究人员认为960 cm-1处出现的谱峰是由于硅氧四面体畸变造成的骨架局部结构不对称所致.42目前认为诱发畸变的一种原因可能是外来客体分子如掺杂的金属离子与载体间的相互作用造成的.在含钨的样品中,此处的谱峰可能是在催化剂制备过程中,部分钨物种进入到了分子筛的骨架中形成的Si―O―W所致.对于WO3/SnO2催化剂,在600-700 cm-1处的谱峰归属于SnO2的Sn―O―Sn振动,43同时与WO3红外谱图相比较,W―O的振动峰发生蓝移,在750及817 cm-1左右出现了吸收峰,可能是由于WO3与SnO2之间强烈的相互作用导致的.结合前面紫外可见漫反射表征可以推测,由于钨物种与载体之间的相互作用,部分钨物种进入到载体晶格中,形成孤立[WO4]四面体钨物种和低聚态的钨物种.

图5 SnO2、WO3和不同载体负载催化剂的FTIR图谱Fig.5 FTIR spectra of SnO2,WO3,and catalysts with different supports
3.2 活性测试结果
3.2.1 不同催化剂对己二酸合成的催化活性
图6是不同催化剂的活性测试结果.从图中可以明显看出,己二酸的得率与催化剂的载体密切相关.以SBA-15为载体时,己二酸的得率只有53.7%,当以HMS为载体时,己二酸得率增至64.3%,但是以SnO2为载体时,催化剂表现出最好的催化活性,己二酸的得率高达96.5%.结合前面的表征结果可知,SnO2与WO3之间存在着强烈的相互作用,其可以抑制WO3晶粒的生长,并使WO3高度分散在催化剂表面,而钨物种的高度分散能够提供更多的活性中心,有利于催化反应的进行,这也是WO3/SnO2催化剂活性高的主要原因.通过对催化剂中O 1s分峰处理得知,WO3/SBA-15和WO3/HMS表面只存在OH氧物种,而在WO3/SnO2中还存在晶格氧,这可能也是该催化剂具有高催化活性的原因之一;并且在WO3/SnO2催化剂中还存在[WO4]四面体和低聚态的W物种,由于WO3/HMS的催化活性比WO3/SBA-15的高,而通过UV-Vis DRS表征结果可知,前者的[WO4]四面体和低聚态的W物种的含量比后者多,从而推测孤立[WO4]四面体和低聚态的W物种也可能是催化剂的活性中心,促进了反应的进行.39此外,SBA-15和HMS为载体的催化剂,活性较差的原因可能是部分钨物种在制备过程中进入到载体孔道中,不能起到很好的催化作用,另一方面是钨物种与载体氧化硅在反应体系中生成了钨硅酸物种.之前已有报道称,44与钨同一主族的钼的氧化物负载在以SiO2为载体的催化反应体系中,钼物种与载体SiO2在有水的条件下,原位生成几种钼硅杂多酸物种,其中一些钼硅物种的形成,降低了催化剂的催化活性.因此,这也解释了钨物种负载在全硅分子筛载体上催化剂活性较低的原因.另外,由于SBA-15和HMS的比表面积很大,重量很轻,因此在相同钨用量的前提下,全硅分子筛的催化剂在无溶剂的反应体系中不能很好地分散,影响了传质速率,从而影响其催化活性.

图6 不同催化剂的催化活性Fig.6 Catalytic performance of different catalysts
3.2.2 WO3/SnO2催化剂的循环使用
为了进一步验证钨锡之间的强相互作用,我们对各种载体催化剂的稳定性也做了研究,催化剂的循环使用次数见图7.从图7可以看出,对于WO3/SnO2催化剂而言,随着反应次数的增加,己二酸的得率缓慢地降低,催化剂在不经任何化学处理的情况下,可重复使用6次以上.第六次反应后己二酸的得率仍保持在80%以上,说明以SnO2为载体的催化剂具有良好的反应稳定性.并且催化剂经简单焙烧后,其催化活性基本可以恢复到其初始性能,推测催化剂失活的可能原因是活性钨物种团聚或被有机物覆盖.而对于WO3/HMS和WO3/SBA-15催化剂,仅仅套用两次活性就明显降低,即使经过焙烧处理后,其活性仍然很低,说明载体与钨物种之间相互作用很弱,催化剂稳定性很差.催化剂失活的主要原因应该是钨物种的流失.为了验证这一推测,我们在反应进行了4 h后,将催化剂过滤除去,滤液继续反应.结果发现,对于WO3/SnO2催化剂,反应几乎完全停止,然而对于WO3/SBA-15和WO3/HMS催化剂体系,反应仍在继续进行.这一现象说明了WO3与SnO2之间存在强烈的相互作用,反应过程中钨物种没有溶脱,而对于WO3/SBA-15和WO3/HMS,固体催化剂过滤除去后,反应仍在继续进行,说明溶液中存在活性均相钨物种,催化剂失活的主要原因是钨物种的流失.

图7 不同催化剂的稳定性测试Fig.7 Stability test of different catalysts
鉴于此,以环氧环己烷为原料,过氧化氢水溶液为氧源,具有高活性高稳定性的WO3/SnO2作为催化剂的己二酸绿色合成工艺,具有良好的实际应用前景.该路线原料易得,生产过程洁净,无有机溶剂和相转移催化剂,不产生氮氧化物等污染物,如大规模工业化可以改变目前工业己二酸生产工艺污染严重的状况,并且多相催化剂分离简单,产物易提纯,为己二酸的新型绿色工艺开发提供了一条新颖的方法.
4 结论
通过简单的浸渍法合成了不同载体的钨基催化剂,并应用到己二酸合成反应中.催化活性结果显示,当以SnO2为载体时,钨基催化剂的催化活性最好,己二酸的得率高达96%以上.而以SBA-15和HMS为载体时,催化剂的催化活性较差,说明载体对催化剂的催化性能有重要的影响.通过表征结果可得知,以SnO2为载体的WO3/SnO2催化剂中氧化钨物种结晶程度较低,粒径很小,并且高度分散在催化剂表面,这是催化剂具有高活性的主要原因.经过稳定性实验发现,WO3/SnO2可以重复使用6次,并且己二酸的得率仍保持在80%以上,说明钨物种与SnO2载体之间存在强烈的相互作用,使催化剂具有高稳定性,为己二酸的新型绿色合成工艺开发提供了一条新途径.
(1)Yang,X.L.;Yin,A.Y.;Dai,W.L.;Fan,K.N.Acta Phys.-Chim.Sin.2011,27(1),177.[杨新丽,尹安远,戴维林,范康年.物理化学学报,2011,27(1),177.]doi:10.3866/PKU.WHXB20110105
(3)Yang,X.L.;Dai,W.L.;Chen,H.;Cao,Y.;Li,H.X.;He,H.Y.;Fan,K.N.J.Catal.2005,229,259.
(4)Sels,B.F.;Devos,D.E.;Jacobs,P.A.Angew.Chem.Int.Edit.2005,44(2),310.
(5)Wilson,R.D.;Barton,D.G.;Baertsch,C.D.;Iglesia,E.J.Catal.2000,194(2),175.doi:10.1006/jcat.2000.2942
(6)Engweiler,J.;Harf,J.;Baiker,A.J.Catal.1996,159(2),259.doi:10.1006/jcat.1996.0087
(7)Sivula,K.;Formal,F.L.;Grätzel,M.Chem.Mater.2009,21,2862.doi:10.1021/cm900565a
(8)Ham,D.J.;Phuruangrat,A.;Thongtem,S.;Lee,J.S.Chem.Eng.J.2010,165,365.doi:10.1016/j.cej.2010.09.003
(9)Takehara,K.;Yamazaki,K.;Miyazaki,M.;Yamada,Y.;Ruenphet,S.;Jahangir,A.;Shoham,D.;Okamura,M.;Nakamura,M.Virus Res.2010,151,102.doi:10.1016/j.virusres.2010.03.006
(10)Abe,R.;Takami,H.;Murakami,N.;Ohtani,B.J.Am.Chem.Soc.2008,130,7780.doi:10.1021/ja800835q
(11)Qamar,M.;Gondal,M.A.;Yamani,Z.H.Catal.Commun.2010,11,768.doi:10.1016/j.catcom.2010.02.012
(12)Morales,W.;Cason,M.;Aina,O.;Tacconi,N.R.;Rajeshwar,K.J.Am.Chem.Soc.2008,130,6318.doi:10.1021/ja8012402
(13)Huang,L.Y.;Xu,H.;Li,Y.P.;Li,H.M.;Cheng,X.N.;Xia,J.X.;Xu,Y.G.;Cai,G.B.Dalton Trans.2013,42,8606.doi:10.1039/c3dt00115f
(14)Li,F.B.;Gu,G.B.;Li,X.J.;Wan,H.F.Acta Phys.-Chim.Sin.2000,16(11),997.[李芳柏,古国榜,李新军,万洪富.物理化学学报,2000,16(11),997.]doi:10.3866/PKU.WHXB20001108
(15)Horsley,J.A.;Wachs,I.E.;Brown,J.M.;Via,G.H.;Hardcastle,F.D.J.Phys.Chem.1987,91(15),4014.doi:10.1021/j100299a018
(16)Engweiler,J.;Harf,J.;Baiker,A.J.Catal.1996,159(2),259.doi:10.1006/jcat.1996.0087
(17)Hilbrig,F.;Göbel,H.E.;Knözinger,H.;Schmelz,H.;Lengeler,B.J.Phys.Chem.1991,95(18),6973.doi:10.1021/j100171a046
(18)Colque,S.;Payen,E.;Grange,P.J.Mater.Chem.1994,4(8),1343.doi:10.1039/jm9940401343
(19)Kim,D.S.;Ostromecki,M.;Wachs,I.E.J.Mol.Catal.A:Chem.1996,106(1-2),93.doi:10.1016/1381-1169(95)00186-7
(20)Kim,D.S.;Ostromecki,M.;Wachs,I.E.;Kohler,S.D.;Ekerdt,J.G.Catal.Lett.1995,33(3-4),209.doi:10.1007/BF00814225
(21)Zhu,Q.J.;Chu,X.F.;Zhang,Z.Y.;Dai,W.L.;Fan,K.N.Appl.Catal.A:Gen.2012,435-436,141.
(22)Mallesham,B.;Sudarsanam,P.;Raju,G.;Reddy,B.M.Green Chem.2013,15(2),478.doi:10.1039/c2gc36152c
(23)Klepel,O.;Böhlmann,W.;Ivanov,E.B.;Riede,V.;Papp,H.Microporous Mesoporous Mat.2004,76(1-3),105.doi:10.1016/j.micromeso.2004.07.038
(24)Koo,D.H.;Kim,M.;Chang,S.Org.Lett.2005,7(22),5015.doi:10.1021/ol052019i
(25)Niu,X.S.;Liu,Y.L.;Hu,P.;Xu,J.Q.Electron.Comp.Mater.2002,21(1),10.[牛新书,刘艳丽,胡 平,徐甲强.电子元件与材料,2002,21(1),10.]
(26)Kamata,K.;Yonehara,K.;Sumida,Y.;Hirata,K.;Nojima,S.;Mizuno,N.Angew.Chem.Int.Edit.2011,50(50),12062.doi:10.1002/anie.v50.50
(27)Wang,J.M.Chem.Technol.Market 2010,33(11),1.[汪家铭.化工科技市场,2010,33(11),1.]
(28)Penate,I.Q.;Lesage,G.;Cognet,P.;Poux,M.Chem.Eng.J.2012,200-202,357.
(29)Wei,L.;Chen,M.;Liu,N.;Wang,S.J.;Wang,J.F.J.Dalian Polytech.University 2010,29(3),216.[魏 莉,陈 梅,刘娜,王少君,王吉峰.大连工业大学学报,2010,29(3),216.]
(30)Jiang,H.;Gong,H.;Yang,Z.H.;Zhang,X.T.;Sun,Z.L.;Kinet,R.Catal.Lett.2002,75(2),315.doi:10.1023/A:1015207214720
(31)Cheng,C.Y.;Lin,K.J.;Prasad,M.R.;Fu,S.J.;Chang,S.Y.;Shyu,S.G.;Sheu,H.S.;Chen,C.H.;Chuang,C.H.;Lin,M.T.Catal.Commun.2007,No.8,1060.
(32)Bohstrom,Z.;Lattes,I.R.;Holmberg,K.Green Chem.2010,12,1861.doi:10.1039/c0gc00032a
(33)Sheng,X.L.;Zhou,Y.M.;Zhang,Y.W.;Duan,Y.Z.;Xue,M.W.Catal.Lett.2012,142,360.doi:10.1007/s10562-012-0769-5
(34)Stein,A.;Fendorf,M.;Jarvie,T.P.;Mueller,K.T.;Benesi,A.J.;Mallouk,T.E.Chem.Mater.1995,7(2),304.doi:10.1021/cm00050a012
(35)Briot,E.;Piquemat,J.Y.;Vennat,M.;Brégeault,J.M.;Chottard,G.;Manoli,J.M.J.Mater.Chem.2000,10(4),953.doi:10.1039/a908428b
(36)Klepel,O.;Böhlmann,W.;Ivanov,E.B.;Riede,V.;Papp,H.Microporous Mesoporous Mat.2004,76(1-3),105.doi:10.1016/j.micromeso.2004.07.038
(37)Weber,R.S.J.Catal.1995,151(2),470.doi:10.1006/jcat.1995.1052
(38)Iglesia,E.;Barton,D.G.;Soled,S.L.;Miseo,S.;Baumgartner,J.E.;Gates,W.E.;Fuentes,G.A.;Meitzner,G.D.Stud.Surf.Sci.Catal.1996,101,533.doi:10.1016/S0167-2991(96)80264-3
(39)Yang,X.L.;Dai,W.L.;Chen,H.;Xu,J.H.;Cao,Y.;Li,H.X.;Fan,K.N.Appl.Catal.A:Gen.2005,283,1.doi:10.1016/j.apcata.2004.12.029
(40)Ansari,S.G.;Dar,M.A.;Dhage,M.S.;Kim,Y.S.;Ansari,Z.A.;Al-Hajry,A.;Shin,H.S.Rev.Sci.Instrum.2009,80(4),045112-1.doi:10.1063/1.3115222
(41)Chen,C.Y.;Li,H.X.;Davis,M.E.Micropor.Mater.1993,2(1),17.doi:10.1016/0927-6513(93)80058-3
(42)Fu,Z.H.;Yin,D.L.;Xie,Q.J.;Zhao,W.;Lv,A.;Yin,D.H.;Xu,Y.Z.;Zhang,L.X.J.Mol.Catal.A:Chem.2004,208(1-2),159.doi:10.1016/S1381-1169(03)00508-9
(43)Zhu,J.J.;Lu,Z.H.;Aruna,S.T.;Aurbach,D.;Aharon,G.Chem.Mater.2000,12,2557.doi:10.1021/cm990683l
(44)Kotbagi,T.V.;Biradar,A.V.;Umbarkar,S.B.;Dongare,M.K.ChemCatChem 2013,5,1531.doi:10.1002/cctc.v5.6
猜你喜欢
风流一代·经典文摘(2023年5期)2023-05-21 11:42:11
电子测试(2018年18期)2018-11-14 02:30:36
化工管理(2017年24期)2017-03-06 06:38:40
化工管理(2017年5期)2017-03-05 08:28:57
光学精密工程(2016年1期)2016-11-07 09:01:00
中国塑料(2016年1期)2016-05-17 06:13:12
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
中国塑料(2014年3期)2014-10-27 08:26:46
无机化学学报(2014年4期)2014-02-28 17:31:23
