
不同浓度比的硫酸根和氯离子溶液中钝化膜的半导体特性转变机制研究
2014-10-18 05:28夏大海杨丽霞
物理化学学报 2014年8期
夏大海 杨丽霞
(1天津大学材料科学与工程学院,天津 300072;2天津大学,天津市材料复合与功能化重点实验,天津 300072;3Department of Chemical and Materials Engineering,University of Alberta,Edmonton T6G 2V4,Alberta,Canada;4中国地质大学材料与化学学院,武汉 430074)
1 引言
随着国民经济的发展,人类对能源的需求越来越大,但石油、煤炭、天然气等不可再生的化石能源却越来越少,因而近年来对核电、风电、太阳能等新型能源的研究与开发得到了全世界范围内的普遍关注.其中,核电作为一种清洁与经济的能源在世界各发达国家都得到迅猛的发展.根据我国的能源政策及发展规划,在未来的十几年里我国将大力发展核电,《国家核电中长期发展规划(2011-2020年)》指出,到2020年我国的核电发电总量将从现在的900万千瓦增加到5800万千瓦,发电装机总量中核电的比例将从2%提高到4%以上.核电设备设计周期一般为40年,由于长期在高温高压水环境及辐照等苛刻条件下运行,且停电检修期间氧的引入会改变二回路的化学及电化学环境(例如腐蚀电位等),因此二回路部件材料常会发生腐蚀、冲蚀、应力腐蚀、辐照脆化等形式的损伤和失效破坏,进而严重威胁核电设备的安全运行.
800镍基合金作为蒸汽发生器二回路传热管在压水堆核电站中应用广泛,不仅源于其优良的耐点蚀及应力腐蚀性能,更主要是由于其在服役环境中能自钝化,即表面形成一层厚度为几纳米至几百纳米(主要取决于温度)的钝化膜.1-3但是,由于二回路长期处于高温高压的环境,一些杂质离子如硫酸根离子和氯离子等4,5会积聚在传热管与管板的缝隙处,使局部环境极具破坏性,其严重程度主要取决于离子的浓度、6局部pH值7以及800合金的腐蚀电位5等.此外,停电检修期间二回路处于室温条件下,而且氧的引入使得800合金的腐蚀电位升高,导致氯离子和硫酸根离子对合金材料的破坏作用增强.初步研究结果表明氯离子和硫酸根离子的浓度对钝化膜的耐蚀性有较大影响,8-12硫酸根离子或氯离子浓度的增加均会使钝化电流密度增大,即钝化膜的溶解速率增大.已有研究主要采用动电位极化曲线和电化学阻抗谱等手段,着重于离子浓度对钝化膜耐蚀性的影响.关于硫酸根离子与氯离子浓度比对钝化膜性质的影响鲜见报道.
事实上,800合金表面的钝化膜是一层具有很高缺陷浓度的半导体膜.根据Sato13提出的钝化膜的能带模型,p型半导体钝化膜的击穿方式为过钝化溶解,而n型半导体的击穿方式则以局部击穿为主,主要表现为点蚀.因此,可以根据钝化膜的半导体类型来深入研究离子浓度比对钝化膜击穿方式的影响,进而阐述离子吸附、缺陷类型与半导体特性的关系.关于钝化膜半导体特性的研究目前普遍采用Mott-Schottky电容分析14-16以及光电化学方法.14另外,还可结合电化学阻抗谱、2,7,17电化学噪声1,18等技术判断钝化膜的耐蚀性,并应用微区电化学技术如扫描电化学显微镜(SECM)、扫描参比电极等12,18-22研究钝化膜的表面电流和电位分布等等.本文利用Mott-Schottky、电化学阻抗谱(EIS)、扫描电化学显微镜等实验技术研究了800合金在含有不同浓度比的硫酸根离子和氯离子溶液中的钝化膜的半导体特性、钝化膜的耐蚀性及其击穿方式,进而提出钝化膜的半导体转变机制,为优化二回路的水化学提供科学依据.
2 实验部分
2.1 实验材料与介质
实验材料为外径15.88 mm、厚1.13 mm的800合金管,其成分如表1所示.将合金管切割成长度为25 mm的短管,然后切割成4个相等的部分,将外表面依次用400#、600#、800#、1200#水砂纸打磨,依次用蒸馏水、乙醇、丙酮清洗,吹干,置于干燥器中干燥24 h.电化学测试介质所用的硫酸钠和氯化钠均由天津市江天化工技术有限公司提供,分析纯级别.
2.2 电化学测试
电化学测试在Gamry(PC-750)电化学工作站进行.阳极极化曲线的扫描速率为0.1667 mV·s-1,电流达到1 mA时终止实验,然后用扫描电镜(SEM)观察表面形貌.EIS测试采用三电极体系,辅助电极为铂金电极,参比电极为饱和甘汞电极(SCE),工作电极为800合金.测试前试样在腐蚀电位下浸泡12 h以形成稳定的钝化膜.EIS测试中施加的正弦扰动幅值为10 mV,扫描频率范围为100 kHz-10 mHz,对数扫频、每倍频程扫点8个,共测56点.EIS数据采用动力学模型进行拟合.

表1 800合金的化学成分(w,%)Table 1 Chemical compositions of Alloy 800(w,%)
钝化膜的半导体特性用Mott-Schottky关系式来描述:
对于n型半导体,
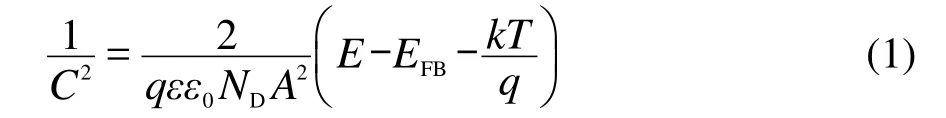
对于p型半导体,

其中,C为空间电荷层电容,ε0是真空介电常数,ε为半导体介电常数,q为电子电荷,ND和NA分别为施主和受主浓度,A为试样面积,E为所施加的电位,EFB为平带电位,k为玻尔兹曼常数,T为绝对温度.由此测量不同电位下的空间电荷层电容C,并通过1/C2对电位E作图,可判定半导体的类型.在Mott-Shottky实验中,电位扫描速率为50 mV·s-1,频率设为1 kHz,扫描的电位区间为合金的钝化区.
2.3 扫描电化学显微镜
扫描电化学显微镜(CHI公司,美国)所用的800合金试样用铜导线焊接,然后封装在环氧树脂中,用抛光机经400#、600#、1200#水砂纸磨平,抛光至光亮,露出合金的外表面作为测试面.SECM所用超微电极为外层包覆玻璃的10 μm的Pt丝.针尖前端的Pt丝与玻璃的直径比约等于5.参比电极为Ag/AgCl电极,对电极为Pt丝电极.所用氧化还原介质为0.9 mmol·L-1二茂铁甲醇,然后加上不同浓度的电解质.SECM采用恒距离模式.首先根据循环伏安曲线,确定超微电极上二茂铁甲醇的氧化电位为0.5 V(vs Ag/AgCl),然后使超微电极维持在此电位,将超微电极移近所测金属表面,进行二维电流扫描,从而获得SECM图像,具体实验过程可参照作者的近期工作.7
3 结果与讨论
3.1 极化曲线
图1为800合金在含有不同浓度比的硫酸根离子、氯离子溶液中的阳极极化曲线以及极化后电极
3.2 电化学阻抗谱分析
为了进一步研究离子浓度比对钝化膜特性的影响,我们采用电化学阻抗谱研究了钝化膜的耐蚀性.图2(a)为800合金在不同离子浓度比的溶液中的Nyquist图.不难发现,阻抗谱均呈现一个不完整的容抗弧特征,这与金属钝化膜EIS的典型特征一致.由于钝化金属的阻抗与电极过程的时间常数较大,因此在所测频率范围内很难呈现完整的阻抗谱图.从图2(a)中可以看到,随着离子浓度比的降低,容抗弧半径减小,这表明钝化膜的耐蚀性降低.图2(b)为800合金在不同离子浓度比的溶液中的Bode图,包括相位角和阻抗的模值随时间的变化.一般从相位角的变化趋势可以看出该电化学体系的时间常数个数,但是对于钝化膜来讲比较困难,可能是由于几个时间常数的大小相近,因此重叠在一起,不易判断.从图中可以看到,随着离子浓度比的降低,相位角曲线向高频方向移动,而高频阻抗模值减小.
对于钝化金属阻抗数据的模型,按照模型的种的表面形貌.由图1可知,合金800在所测溶液中均为自钝化,钝化电流密度小于1 μA·cm-2,表明钝化膜的溶解速度很低.随着硫酸根离子与氯离子浓度比的降低,钝化电流密度略有升高,钝化电位区间减小.另外,值得注意的是,钝化膜击穿时电流值的增大方式是不同的:当硫酸根离子与氯离子的浓度比较高时,其极化电流值逐渐增加;当硫酸根离子与氯离子的浓度比较低时,其极化电流值突然增加.Sato13也发现类似的现象:从Fe在硫酸中钝化膜为n型半导体,通过观察其在硫酸中的阳极极化曲线可以看到,钝化膜击穿时电流值突然增大,而Ni在硫酸中的钝化膜表现为p型半导体特征,通过观察其在硫酸中的阳极极化曲线发现,钝化膜击穿时电流值逐渐增大.当浓度比较高时,以过钝化溶解为主;当浓度比较低时,钝化膜易发生点蚀.从电极表面极化后的SEM形貌可以看到,随着浓度比的降低,电极表面由过钝化溶解转变为明显的点蚀特征.当浓度比较高时,电极表面无明显孔蚀的特征,主要是以过钝化溶解为主(如图1(b,c,d)所示);而当浓度比较低如氯离子的浓度增加到0.1 mol·L-1时,电极表面的孔蚀特征明显(如图1(e,f)所示).我们初步认为,腐蚀形态(孔蚀或过钝化)的改变与钝化膜的半导体特性有关,因为钝化膜的半导体特性会影响钝化膜的击穿方式.稍后将做详细分析.类,大概可以分为:(1)电化学等效电路模型;23-27(2)点缺陷模型(PDM);28-31(3)动力学模型.1,32,33电化学等效电路模型是目前广泛采用的一种方法,但是对于等效电路的选择,不同学者有不同的看法.23-27点缺陷模型是MacDonald等28-31发展的一种新方法,针对钝化膜中各种缺陷反应的动力学方程式,对阻抗数据进行拟合,取得了较为满意的结果,但最近Marcus等34,35认为该模型有缺点,他们提出了一个更为适用的物理模型.动力学模型则是根据所研究电化学体系的状态变量,直接推导得到体系的阻抗的表达式,较为可靠,因此本文采用动力学模型对EIS数据进行解析.根据动力学模型,在线性范围内如果电极受到一个ΔE电位的扰动,钝化膜的厚度变化为Δl,此时的法拉第电流为if,钝化膜的溶解电流为is,则在有限的时间范围内,钝化膜的厚度l随时间t的变化关系式为:1
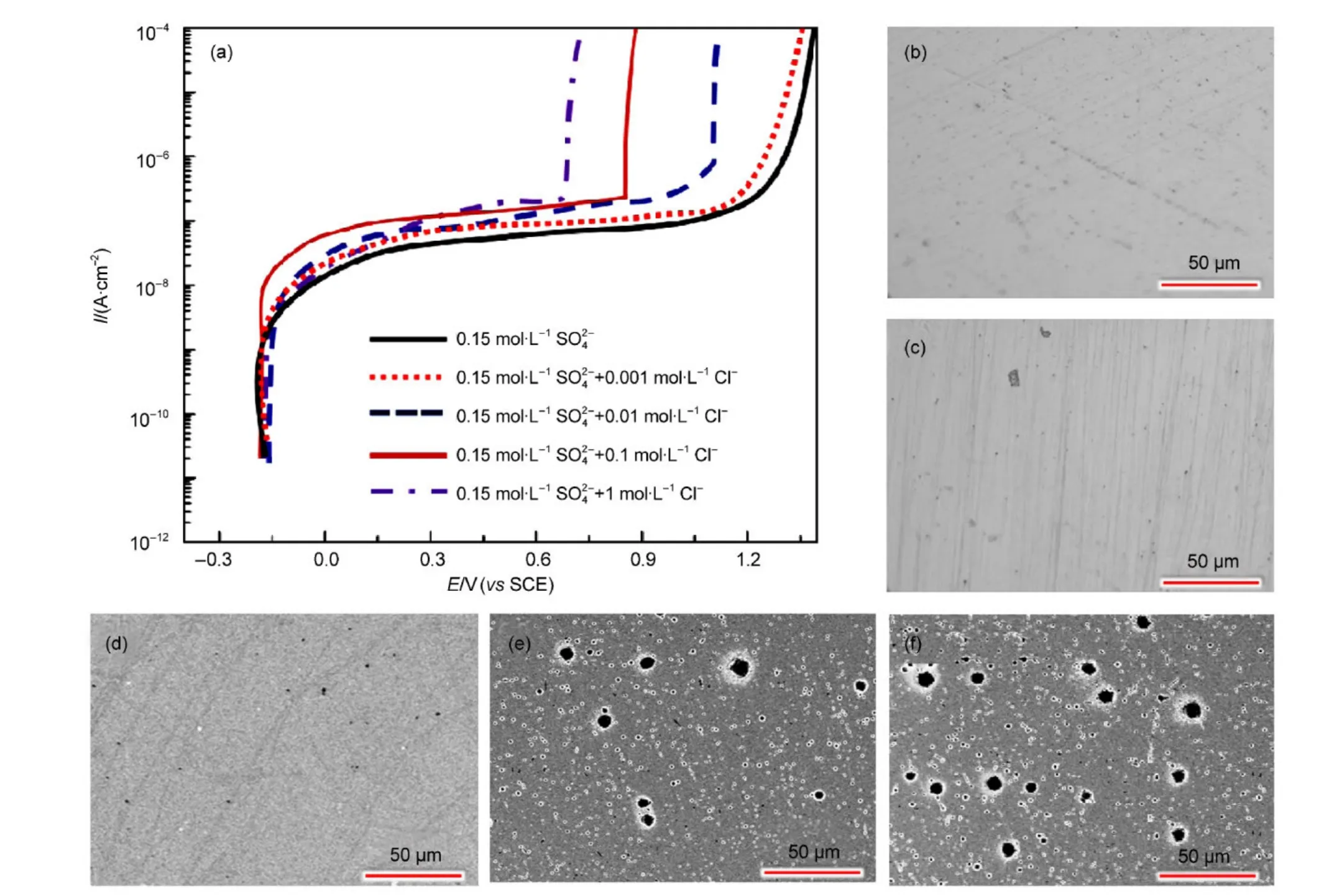
图1 800合金在含有不同离子浓度比的硫酸根和氯离子溶液中的极化曲线和极化后的SEM图Fig.1 Polarization curves in solutions with different concentration ratios of sulfate to chloride and SEM images after the polarization test
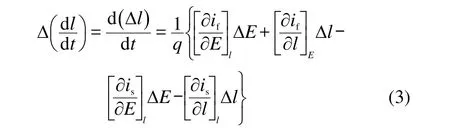
其中q为电子电荷.动力学模型认为钝化膜的极化电阻Rp约等于钝化膜的溶解电阻Rs,即

图2(c)为得到的极化电阻Rp值随着氯离子浓度的变化情况.不难发现,极化电阻随着离子浓度比的降低而减小,说明氯离子在钝化膜表面的吸附导致钝化膜的溶解速度增加.与不含氯离子的极化电阻相比,含有1 mol·L-1氯化钠溶液的极化电阻值降低了一半.
3.3 Mott-Schottky分析

图2 800合金在含有不同离子浓度比的溶液中的EIS图和动力学模型拟合结果Fig.2 EIS plots ofAlloy 800 in solutions with different ion concentration ratio of sulfate to chloride and fitting results using kinetic model

图3 Mott-Schottky曲线(a)以及线性段斜率值(b)Fig.3 Mott-Schottky curves(a)and the slopes in linear region(b)
图3(a)为800合金在不同离子浓度比的溶液中的Mott-Schottky曲线.从图中可以看到,在-0.2-0.05 V(vs SCE)的电位区间内,1/C2与电位呈线性关系.超出该电位区间,可能有法拉第电流产生,因此该区域的数据并不可靠.一般认为,正的斜率表明钝化膜为n型半导体,负的斜率表明钝化膜为p型半导体;且直线段斜率的绝对值与钝化膜的缺陷浓度有关,绝对值越小,表明钝化膜的缺陷浓度越大.在本文研究中,随离子浓度比的降低,线性斜率由负值变为正值且线性斜率绝对值不断减小,说明半导体特性随着离子浓度比的降低发生明显转变.离子浓度比对钝化膜的半导体特性的影响可分为两个阶段:(1)当离子浓度比较高(如氯离子的浓度小于0.1 mol·L-1)时,钝化膜为p型半导体,随离子浓度比的降低钝化膜中的施主浓度增加.这与文献1中的报道是一致的:在只含硫酸根或者只含硫代硫酸根离子的溶液中,800合金表面的钝化膜为p型半导体,此时钝化膜中的空位类型主要是金属空位为主(主要是Fe,Ni,Cr的金属空位).(2)当离子浓度比较低(如氯离子的浓度大于0.1 mol·L-1)时,钝化膜的半导体特性由p型转变为n型,钝化膜中的主要缺陷由施主变为受主,而且缺陷浓度继续减小.文献1中也指出在只含氯离子或者同时含有0.6 mol·L-1氯离子+0.075 mol·L-1硫代硫酸根离子的溶液中(此时氯离子的吸附占主导),800合金表面的钝化膜为n型半导体,此时钝化膜中的空位类型主要是氧空位.从图3(b)可以明显看出Mott-Schottky曲线直线段的斜率值随离子浓度比的变化规律,随着浓度比的降低,斜率从负值变为正值.本文并没有计算缺陷浓度的值,因为钝化膜的成分不是简单的单一的氧化物,而且众多研究表明钝化膜中含有氢氧化物,36而氢氧化物的介电常数是未知的.
3.4 扫描电化学显微镜
图4给出了800合金在不同离子浓度比的溶液中的SECM图.图中的电流值为Fc的还原电流.电流值的大小与钝化膜的电化学活性(或者溶解速度)正相关,Fc的还原电流值越大,钝化膜的溶解速度越大,如图4(f)所示.图4(a)为只有0.15 mol·L-1硫酸根时的SECM图,图上不同灰色代表钝化膜表面不同的活性,即钝化膜不同的溶解速度.部分区域较大的电流值可能是由于晶界或者夹杂物的影响所致,7,37,38此处的钝化膜较为薄弱,因此活性较高,溶解速度较快.图4(b-e)为添加不同浓度氯离子之后的SECM图.由图可知,随着离子浓度比的降低,钝化膜表面的电流普遍增加,表明钝化膜表面的溶解速度逐渐增大.很显然一些活性较高的点(图中深色区)的溶解速度也随之增加,在较高的电位下,这些活性较高的点将优先被击穿,进而发展成为蚀孔.
3.5 半导体特征转变机制
在上述研究的基础上,我们进一步讨论了800合金钝化膜的半导体特性转变机制,见图5.该体系的电位降主要包括三个部分:金属/钝化膜处的电位降(Φm/f),钝化膜内的电位降(Φf)以及钝化膜/溶液界面的电位降(Φf/s).在本文所研究的体系中(中性溶液),H2O分子会在钝化膜表面发生分解,分解产物氧进入钝化膜,是钝化膜的主要成膜反应;30同时,钝化膜中的金属离子MZ+会溶解进入溶液中,是钝化膜的主要溶解反应.

图4 含不同氯离子浓度的0.15 mol·L-1硫酸钠溶液中800合金的SECM二维电流分布Fig.4 SECM two-dimension current distribution of Alloy 800 in 0.15 mol·L-1 sulfate with different chloride concentrations
在只有硫酸根离子的溶液中,硫酸根离子在钝化膜表面的吸附导致钝化膜中的金属离子溶解,1使金属空位增多,此外H2O分子分解产生的氧容易进入钝化膜内,填补钝化膜内的氧空位,由此钝化膜表现为p型半导体的特征.当少量的氯离子加入到硫酸根溶液后,仍以硫酸根离子的表面吸附占主导,并未改变半导体的特性,此时仍然以金属空位为主.但是,少量氯离子的加入会导致钝化膜/溶液界面处的电势差(Φf/s)增加,使得金属离子的溶解速度增加,最终导致钝化膜中的金属空位增多,如图5(a)所示.随氯离子浓度的增加,氯离子在钝化膜表面的吸附量增大,吸附在氧空位(VO••)上的氯离子使得H2O分子的分解反应难以进行(如反应式(5)),因此导致钝化膜中的氧空位含量增多,28,39此外,钝化膜/溶液界面处的电势差也随氯离子浓度的增加而增加,导致钝化膜的溶解速度增加(如图2的EIS结果所示).当氯离子浓度足够高时,钝化膜的成膜反应达到了另外一个平衡状态,此时钝化膜呈现n型半导体的特征(如图5(b)所示).

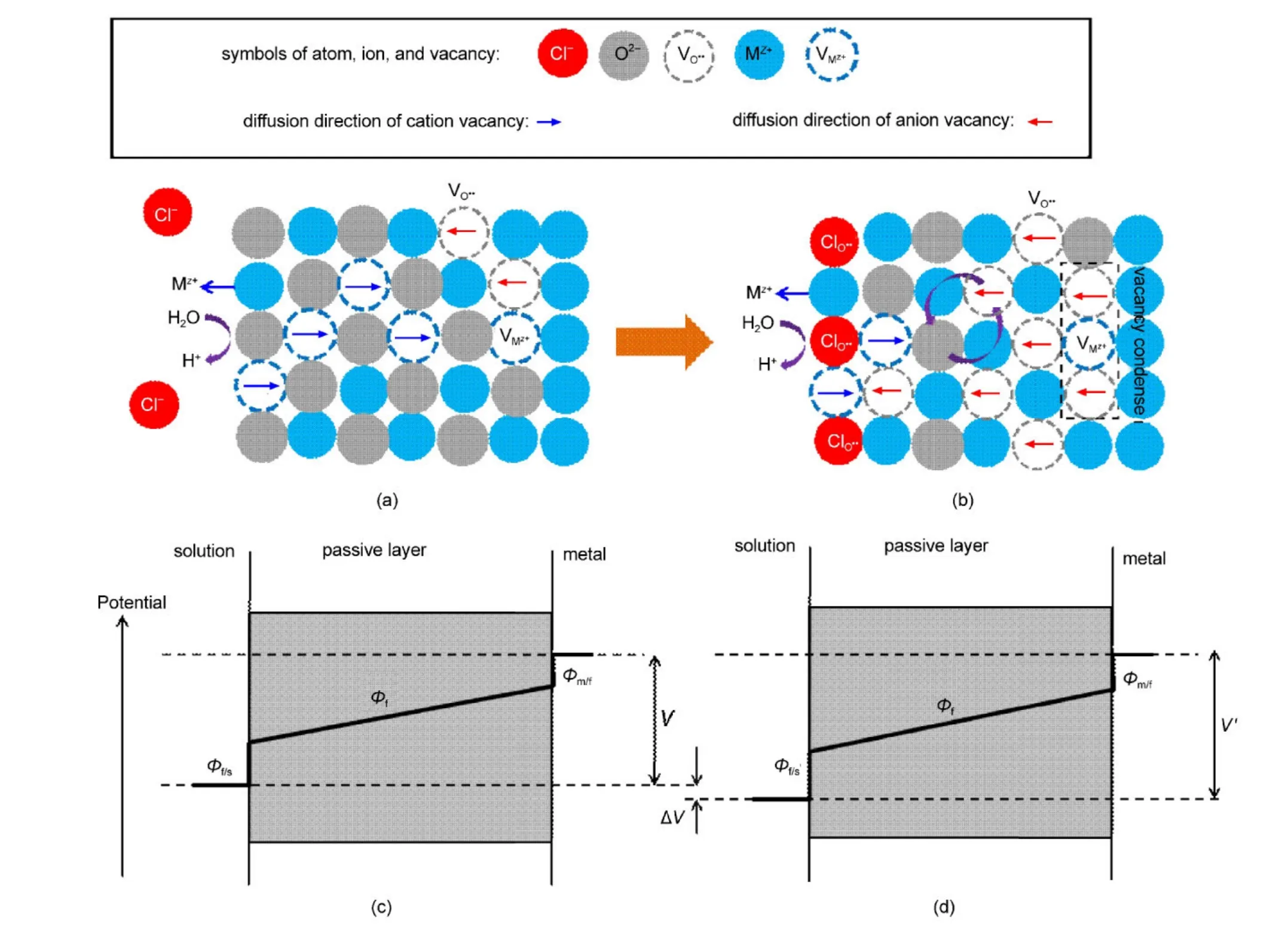
图5 钝化膜的半导体转变示意图Fig.5 Schematic diagram of semiconductivity conversion of passive film
根据Sato13提出的钝化膜的能带模型,p型半导体的击穿方式为过钝化溶解,即钝化膜整体的溶解;n型半导体的击穿方式则为局部的击穿,击穿方式一般表现为孔蚀.可以这样理解:p型半导体的金属空位源于钝化膜的外层,是由钝化膜的溶解产生的,可认为是钝化膜外层的均匀反应,当电位较高时,金属的过钝化容易进行.对于n型半导体,其缺陷主要是氧空位,而氧空位源自金属/钝化膜界面,当氧空位的产生速度大于其消逝速度,多余的氧空位会在金属/钝化膜界面累积,特别容易在一些钝化膜的薄弱处(如晶界处)累积,进而成为点蚀优先发生的地点.
另外有两个问题需要说明:(1)钝化膜的半导体特性与腐蚀形态有关,n型半导体钝化膜发生击穿时,不一定是SEM下可见的形状规则的蚀孔,也有可能是极其不规则甚至看不到的蚀孔,但是其击穿方式可以归纳为钝化膜的局部击穿,从极化曲线上表现为电流值的突然增大;而p型半导体膜发生击穿时,一般对应于电极表面的整体的过钝化溶解.(2)钝化膜的半导体特性与腐蚀速度之间并无直接的联系,也就是说,n型半导体膜的溶解速度不一定比p型半导体的溶解速度大,钝化膜的溶解速度还要取决于介质的种类和介质的浓度等等.本文中,与硫酸根相比,氯离子的侵蚀性较强,因此在氯离子浓度较高的溶液中,钝化膜的溶解速度较快.
4 结论
本文采用电化学阻抗谱、Mott-Schottky电容分析以及扫描电化学显微镜技术方法研究了800合金在不同浓度比的硫酸根与氯离子溶液中的半导体特性,可以得到以下结论:
(1)800合金表面钝化膜的半导体特性由硫酸根与氯离子的浓度比决定,随着浓度比的降低,钝化膜的半导体特性发生转变.
(2)当溶液中硫酸根与氯离子的浓度比较高时,钝化膜为p型半导体;当溶液中硫酸根与氯离子的浓度比较低时,钝化膜为n型半导体.
(3)半导体类型的改变与硫酸根与氯离子的竞争吸附有关,二者的竞争吸附会导致电极/溶液界面电势差改变,进而使钝化膜中的空位类型改变,最终使半导体类型改变.
(4)由极化后电极的SEM形貌可知,随着氯离子浓度的增加,电极表面由过钝化溶解转为明显的点蚀特征,也说明钝化膜的半导体的击穿方式不同,与钝化膜的半导体特性发生转变有关.
(1)Xia,D.H.;Song,S.Z.;Zhu,R.K.;Behnamian,Y.;Shen,C.;Wang,J.H.;Luo,J.L.;Lu,Y.C.;Klimas,S.Electroch imica Acta 2013,111,510.doi:10.1016/j.electacta.2013.08.030
(2)Xia,D.H.;Zhu,R.K.;Behnamian,Y.;Shen,C.;Luo,J.L.;Lu,Y.C.;Klimas,S.Journal of the E lectrochemical Society 2014,161,C201.
(3)Zhu,R.K.;Lu,B.T.;Luo,J.L.;Lu,Y.C.Applied Surface Science 2013,270,755.doi:10.1016/j.apsusc.2013.01.150
(4)Lu,B.T.;Luo,J.L.;Lu,Y.C.Journal of the Electrochemical Society 2007,154,C379.
(5)Lu,B.T.;Luo,J.L.;Lu,Y.C.Electrochimica Acta 2008,53,4122.doi:10.1016/j.electacta.2007.12.070
(6)Lu,Y.AE CL Nuclear R eview 2012,1,13.
(7)Luo,B.;Xia,D.H.A cta Ph ysico-Ch imica Sinica 2014,30,59.[罗 兵,夏大海.物理化学学报,2014,30,59.]doi:10.3866/PKU.WHXB201311221
(8)Montemor,M.F.;Ferreira,M.G.S.;Walls,M.;Rondot,B.;Belo,M.C.Corrosion 2003,59,11.doi:10.5006/1.3277531
(9)Huang,J.;Wu,X.;Han,E.H.Corrosion S cience 2009,51,2976.doi:10.1016/j.corsci.2009.08.002
(10)Huang,J.;Wu,X.;Han,E.H.Corrosion Science 2010,52,3444.doi:10.1016/j.corsci.2010.06.016
(11)Kim,H.;MacDonald,D.D.Corrosion Science 2010,52,1139.doi:10.1016/j.corsci.2009.12.006
(12)Meng,F.;Han,E.H.;Wang,J.;Zhang,Z.;Ke,W.E lectrochimica Acta 2011,56,1781 doi:10.1016/j.electacta.2010.08.028
(13)Sato,N.Journal of the Electrochemical Society 1982,129,255.doi:10.1149/1.2123808
(14)Lu,B.T.;Luo,J.L.;Lu,Y.C.Electrochimica A cta 2013,87,824.doi:10.1016/j.electacta.2012.10.006
(15)Lu,B.T.;Tian,L.P.;Zhu,R.K.;Luo,J.L.;Lu,Y.C.E lectrochimica Acta 2011,56,1848.doi:10.1016/j.electacta.2010.09.104
(16)Yang,M.Z.;Luo,J.L.;Yang,Q.;Qiao,L.J.;Qin,Z.Q.;Norton,P.R.Journal o f the Electrochemical Society 1999,146,2107.doi:10.1149/1.1391899
(17)Li,J.;Xu,Z.Y.;Li,J.Y.;Jiao,D.Acta Physico-Chimica Sinica 2010,26,2638.[李 进,许兆义,李久义,焦 迪.物理化学学报,2010,26,2638.]doi:10.3866/PKU.WHXB20100927
(19)Maurice,V.;Marcus,P.E lectrochimica Acta 2012,84,129.doi:10.1016/j.electacta.2012.03.158
(20)Massoud,T.;Maurice,V.;Klein,L.H.;Wiame,F.;Marcus,P.Corrosion Science 2013,69,245.doi:10.1016/j.corsci.2012.12.010
(21)Massoud,T.;Maurice,V.;Klein,L.H.;Marcus,P.Journal of the E lectrochemical Society 2013,160,C232.
(22)Tian,H.W.;Li,W.H.;Wang,D.P.;Hou,B.R.Acta P hysico-Chimica Sinica 2012,28,137.[田惠文,李伟华,王大鹏,侯保荣.物理化学学报,2012,28,137.]doi:10.3866/PKU.WHXB201228137
(23)Xia,D.;Wang,J.;Jiang,Y.;Li,N.;Zhou,C.Journal of Tianjin University(Science and Technology)2013,46,503.
(24)Staehle,R.W.;Gorman,J.A.Corrosion 2004,60,115.doi:10.5006/1.3287716
(25)Xia,D.H.;Song,S.Z.;Wang,J.H.;Bi,H.C.CIESC Journal 2012,63,1797.
(26)Xia,D.H.;Song,S.Z.;Wang,J.H.;Bi,H.C.Transactions of Tianjin University 2012,18,15.
(27)Zheng,X.;Xia,D.;Wang,H.;Fu,C.Anti-Corrosion Methods and Materials 2013,60,153.doi:10.1108/00035591311315382
(28)Zhang,Y.;Urquidi-MacDonald,M.;Engelhardt,G.R.;MacDonald,D.D.Electroch imica Acta 2012,69,1.doi:10.1016/j.electacta.2012.01.022
(29)Zhang,Y.;Urquidi-MacDonald,M.;Engelhardt,G.R.;MacDonald,D.D.Electroch imica Acta 2012,69,12.doi:10.1016/j.electacta.2012.01.023
(30)Zhang,Y.;Urquidi-MacDonald,M.;Engelhardt,G.R.;MacDonald,D.D.Electroch imica Acta 2012,69,19.doi:10.1016/j.electacta.2012.01.024
(31)Mao,F.;Sharifi-Asl,S.;Yu,J.;MacDonald,D.D.Journal of the E lectrochemical Society 2014,161,C254.
(32)Shao,H.B.;Wang,J.M.;He,W.C.;Zhang,J.Q.;Cao,C.N.E lectrochemistry Communications 2005,7,1429.doi:10.1016/j.elecom.2005.10.002
(33)Shao,H.B.;Wang,J.M.;Wang,X.Y.;Zhang,J.Q.;Cao,C.N.E lectrochemistry Communications 2004,6,6.doi:10.1016/j.elecom.2003.10.007
(34)Leistner,K.;Toulemonde,C.;Diawara,B.;Seyeux,A.;Marcus,P.Journal of the E lectrochemical Society 2013,160,C197.
(35)Seyeux,A.;Maurice,V.;Marcus,P.Journal of the E lectrochemical Society 2013,160,C189.
(36)Zhang,C.S.;Luo,J.L.;Munoz-Paniagua,D.;Norton,P.R.T hin Solid Films 2006,503,149.doi:10.1016/j.tsf.2005.10.083
(37)Zhu,R.K.;Nowierski,C.;Ding,Z.F.;Noel,J.J.;Shoesmith,D.W.Chemistry of Materials 2007,19,2533.doi:10.1021/cm070023d
(38)Zhu,R.K.;Qin,Z.Q.;Noel,J.J.;Shoesmith,D.W.;Ding,Z.F.A nalytical Chemistry 2008,80,1437.doi:10.1021/ac701796u
(39)Sharifi-Asl,S.;Taylor,M.L.;Lu,Z.;Engelhardt,G.R.;Kursten,B.;MacDonald,D.D.Electrochimica Acta 2013,102,161.doi:10.1016/j.electacta.2013.03.143
猜你喜欢
建筑材料学报(2022年3期)2022-03-29
世界有色金属(2021年6期)2021-06-14
陶瓷学报(2019年5期)2019-01-12
厦门理工学院学报(2016年1期)2016-12-01
铁道科学与工程学报(2016年10期)2016-11-12
安徽工程大学学报(2016年2期)2016-07-20
浙江大学学报(工学版)(2016年2期)2016-06-05
中国水利(2015年21期)2015-01-30
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
