
新型藤黄酸衍生物的合成及其抗肿瘤活性*
2014-08-30 09:10:54黎金海张志佳陶移文廖思燕
合成化学 2014年6期
黎金海,黄 雁,谭 茵,张志佳,陶移文,廖思燕
(1.广州医科大学 a.药学院;b.附属第三医院,广东 广州 510182)
·研究论文·
新型藤黄酸衍生物的合成及其抗肿瘤活性*
黎金海1a,1b,黄 雁1a,谭 茵1a,张志佳1a,陶移文1a,廖思燕1a
(1.广州医科大学 a.药学院;b.附属第三医院,广东 广州 510182)
以藤黄酸(1)为原料,分别与HBr和有机胺反应,合成了9个新型的藤黄酸衍生物(2~6),其结构经1H NMR,MS和HR-MS表征。采用MTT法测定了2~6对人结肠腺癌细胞(RKO)、人肝癌细胞(HepG-2)和人卵巢腺癌细胞(OVCAR-3)的体外抗肿瘤活性。结果表明,藤黄酸(N-丙基对甲苯磺酰胺)酯3,藤黄酸(N-丙基苯丙酰胺)酯4和N-色胺藤黄酰胺6b的抗肿瘤活性显著高于1;33-羟基转位藤黄酸2的抗肿瘤活性则大大降低。
藤黄酸;衍生物;合成;抗肿瘤活性
藤黄酸(1)是由藤黄科植物藤黄树分泌的树脂中提取出来的一种天然化合物[1],具有较高的抗肿瘤活性和良好的细胞选择性[2]。1对癌细胞有选择性杀灭效果,而对正常的造血系统和白细胞没有影响[3-4]。研究表明,1对肝癌[5-6]、白血病[7-8]和骨肉瘤[9]等多种肿瘤细胞都有抑制作用。
近年来,为得到高效低毒和水溶性好的1的衍生物,研究人员对其进行了大量的化学结构修饰,主要集中在6-羟基,8-羰基和30-羧基等位点。例如,Zhang S L等[10]、 Zhang X J等[11]、 Li等[12]和Sun H P等[13]分别通过对1的32-位,34-位,37-位和39-位进行结构修饰,合成了一系列1的衍生物。生物活性研究结果表明,1的衍生物的抗肿瘤活性显著强于1;He等[14]通过酯化1的30-羧基,改善了其水溶性,提高了抗肿瘤活性。此外,1的30-羧基可进行酯化或酰胺化等多种修饰方式,通过引进某些合适的基团增强其抗肿瘤活性,而9-位,10-位双键对于1诱导细胞凋亡有重要作用,当双键被还原或者发生迈克尔加成后,抗肿瘤活性大大降低[15-16]。
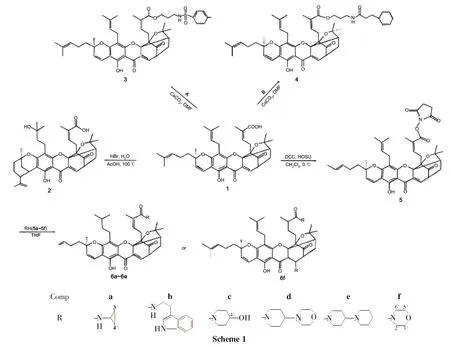
为进一步研究结构修饰对1抗癌活性的影响,本文以1为原料,分别与HBr和有机胺反应,合成了9个新型的藤黄酸衍生物(2~6,Scheme 1),其结构经1H NMR,MS和HR-MS表征。采用MTT法测定了2~6对人结肠腺癌细胞(RKO)、人肝癌细胞(HepG-2)和人卵巢腺癌细胞(OVCAR-3)的体外抗肿瘤活性。结果表明,3,4和6b的抗肿瘤活性显著高于1,2的抗肿瘤活性则大大降低。
1 实验部分
1.1 仪器与试剂
Varian Mercury-Plus 300MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);MAT-95XP型高分辨质谱仪。
1,广州清平中药材市场,使用前经提纯;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1)33-羟基转位藤黄酸(2)的合成
在反应瓶中加入1500mg(0.8mmol)和乙酸15mL,搅拌使其溶解;缓慢滴加35%氢溴酸溶液1mL,滴毕,于100℃(浴温)反应2h。冷却至室温,倾入3倍量的水中,析出乳黄白色固体,用乙醚提取,饱和盐水洗涤,无水硫酸钠干燥,旋蒸除去乙醚,残留物经硅胶柱层析[洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=5∶1]纯化得乳黄色胶体2344mg,产率为69%;1H NMRδ: 12.96(s,1H,6-OH),7.52(d,J=6.6Hz,1H,10-H),5.55(s,1H,27-H),5.20(d,1H,33-OH),4.76~4.67(m,2H,32-H),4.50(brs,1H,40a-H),4.35(brs,1H,40b-H),4.13(m,2H,34-H),3.89(s,3H,35-H),3.51(s,1H,11-H),2.90(m,2H,26-H),2.71(m,2H,31-H),2.49(d,J=9.6Hz,1H,22-H),2.33(s,1H,21a-H),2.16(m,3H,39-H),2.06(s,3H,29-H),1.75(s,3H,35-H),1.66(s,2H,36-H),1.58(s,1H,20b-H),1.40(s,1H,21a-H),1.32~1.30(m,6H,40,19-H);MSm/z: 646.8{[M+H]+};HR-MSm/z:Calcd for C38H46O9[M]646.3136,found 646.3134。
(2)藤黄酸(N-丙基对甲苯磺酰胺)酯(3)和藤黄酸(N-丙基苯丙酰胺)酯(4)的合成
在反应瓶中加入三溴丙胺氢溴酸盐876mg(4mmol)和二氯甲烷20mL,搅拌使其溶解;滴加三乙胺1.2mL,滴毕,搅拌10min,滴加对甲苯酰氯382mg(2mmol),滴毕,搅拌反应4h。滴加0.1mol·L-1盐酸4mL,滴毕,搅拌15min,析出淡黄色固体,过滤得A(Chart 1)498mg,产率85%。
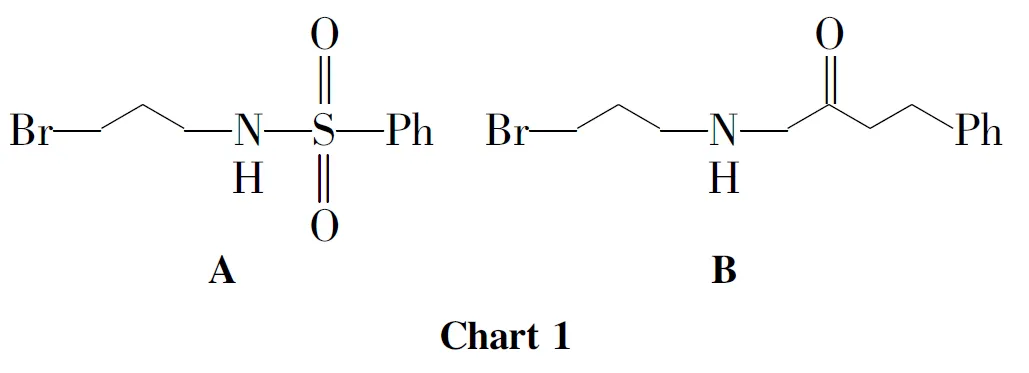
在反应瓶中加入1500mg(0.78mmol),无水碳酸铯400mg(1.36mmol)和DMF 15mL,搅拌使其溶解;搅拌下于室温反应约0.5h,滴加A400mg(1.36mmol)的DMF(15mL)溶液,滴毕,反应24h(TLC检测)。用乙酸乙酯萃取,合并有机相,用饱和食盐水(3×40mL)洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析(洗脱剂: A=3∶1)纯化得淡黄色胶体3530mg,收率63%;1H NMRδ: 12.80(s,1H,6-OH),7.69(d,J=7.0Hz,2H,ArH),7.50(d,J=6.7Hz,1H,10-H),7.26(d,J=8.5Hz,2H,ArH),6.61(d,J=10.1Hz,1H,4-H),5.98(t,J=7.6Hz,1H,27-H),5.41(d,J=10.1Hz,1H,3-H),5.01(m,2H,32,37-H),4.77(q,J=7.1Hz,2H,CO2CH2CH2CH2),3.87(m,2H,CO2CH2CH2CH2),3.48(s,1H,11-H),3.29(m,2H,31-H),2.99(m,3H,Ar-H),2.91(m,2H,26-H),2.50(d,J=9.5Hz,1H,22-H),2.41(s,2H,CO2CH2CH2CH2),2.30(m,2H,21-H),2.04(s,2H,36-H),1.73(s,3H,25-H),1.68(s,3H,29-H),1.64(s,2H,20-H),1.62(s,3H,34-H),1.59(s,6H,35,39-H),1.55(m,3H,40-H),1.41(s,3H,24-H),1.26(s,3H,19-H);MSm/z: 840.7{[M+H]+}。
以3-苯丙酰氯[0.2mL(2mmol)]代替对甲苯酰氯,用类似的方法制得B(Chart 1),进而合成了淡黄色胶体4518mg,收率63%;1H NMRδ: 12.86(s,1H,6-OH),7.49(d,J=6.9Hz,1H,10-H),7.28~7.17(m,5H,ArH),6.60(d,J=10.1Hz,1H,4-H),6.05(t,J=7.6Hz,1H,27-H),5.40(d,J=10.1Hz,1H,3-H),5.03(m,2H,32,37-H),3.99(m,2H,NH),3.88~3.73(m,2H,CO2CH2CH2CH2),3.48(s,1H,11-H),3.30(m,2H,31-H),3.13~3.08(m,2H,26-H),2.96(t,J=7.6Hz,4H,NHCOCH2CH2),2.62(m,1H,22-H),2.48(s,3H,CO2CH2CH2CH2),2.30(m,2H,21-H),2.01(s,2H,36-H),1.72(s,3H,25-H),1.68(s,3H,29-H),1.67(s,2H,20-H),1.64(s,3H,34-H),1.57(s,6H,35,39-H),1.55(m,3H,40-H),1.41(s,3H,24-H),1.28(s,3H,19-H);MSm/z: 818.7{[M+H]+}。
(3)6a~6f的合成(以6a为例)
在反应瓶中加入1500mg(0.8mmol),N-羟基琥珀酰亚胺75mg(HOSu)和干燥CH2Cl220mL,搅拌使其溶解;冰浴(0℃)冷却,搅拌下加入N,N′-二环己基二亚胺(DCC),于室温反应1h;过滤,滤液浓缩,干燥后滴加98%环丙胺5a0.12mL(1.65mmol)的THF(15mL)溶液,滴毕,于室温反应4h。用乙酸乙酯萃取,分出乙酸乙酯层,用饱和食盐水洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析[洗脱剂:V(二氯甲烷)∶V(乙酸乙酯)=10∶1]纯化得黄色胶体N-环丙基藤黄酰胺6a。
分别以色胺(5b)、 4-羟基哌啶(5c)、 4-吗啡啉哌啶(5d)、 4-哌啶基哌啶(5e)和吗啡啉(5f)代替5a,用类似的方法合成了淡黄色胶体N-色胺藤黄酰胺(6b)、N-(4-羟基哌啶)藤黄酰胺(6c)、N-(4-吗啡啉哌啶)藤黄酰胺(6d)、N-(4-哌啶基哌啶)藤黄酰胺(6e)、 10,N-二吗啡啉藤黄酰胺(6f)。
6a:产率48%;1H NMRδ: 12.82(s,1H,6-OH),7.55(d,J=6.7Hz,1H,10-H),6.68(d,J=10.1Hz,1H,4-H),6.76(s,1H,NH),5.46(d,J=10.1Hz,1H,3-H),5.26(t,1H,J=7.5Hz,27-H),5.03(m,2H,32,37-H),3.47(m,1H,11-H),3.30(m,2H,31-H),3.20(m,2H,26-H),2.75~2.53(m,2H,21-H),2.33(d,J=9.6Hz,1H,22-H),2.06(m,2H,36-H),1.96(m,4H,3′,4′-H),1.78(s,3H,25-H),1.73(s,3H,29-H),1.69(s,3H,20-H),1.66(s,3H,34-H),1.60(s,3H,39-H),1.56(s,3H,35-H),1.45(s,3H,40-H),1.35~1.30(m,6H,24,19-H);MSm/z: 684.5{[M+H]+};HR-MSm/z:Calcd for C41H49NO7667.3504,found 667.3499。
6b: 产率50%;1H NMRδ: 12.83(s,1H,6-OH),7.97(s,1H,CONH),7.59(d,J=7.0Hz,1H,10-H),7.39(d,J=7.4Hz,1H,tryptamine-H),7.33(d,J=8.9Hz,1H,tryptamine-H),7.11(m,3H,tryptamine-H),6.99(s,1H,tryptamine-NH),6.65(d,J=10.1Hz,1H,4-H),5.41(d,J=10.1Hz,1H,3-H),5.33(s,1H,27-H),5.03(m,2H,32-H,37-H),3.54(m,1H,CONH),3.48(s,1H,11-H),3.34~3.26(m,2H,31-H),2.98(m,2H,26-H),2.70(m,1H,21a-H),2.47(d,J=9.2Hz,1H,22-H),2.25(m,1H,21b-H),2.04(s,2H,36-H),1.92(s,2H,CONHCH2CH2),1.74(s,3H,25-H),1.70(s,3H,29-H),1.64(s,5H,20,34-H),1.58(s,6H,35,39-H),1.54(m,3H,40-H),1.38(s,3H,24-H),1.17(s,3H,19-H);MSm/z: 771.7{[M+H]+}。
6c: 产率56%;1H NMRδ: 12.81(s,1H,6-OH),7.57(d,J=6.7Hz,1H,10-H),6.69(d,J=10.6Hz,1H,4-H),5.90(s,1H,27-H),5.47(d,J=9.9Hz,1H,3-H),5.08(s,2H,32-H,37-H),4.13(s,1H,4′-OH),3.45(m,1H,11-H),3,31(m,2H,31-H),2.99(m,2H,26-H),2.51(d,J=9.5Hz,1H,22-H),2.35(s,2H,21-H),2.06(m,2H,36-H),1.76(s,6H,25,29-H),1.73(s,6H,20,34-H),1.67(s,6H,35,39-H),1.60~1.57(m,6H,40,24-H),1.46~1.43(m,5H,3′,4′,5′-H),1.39(s,3H,19-H),1.30~1.28(s,3H,19-H);MSm/z: 712.5{[M+H]+};HR-MSm/z:Calcd for C43H53NO8[M]711.3766,found 711.3762。
6d:产率53%;1H NMRδ: 12.85(s,1H,6-OH),7.63(d,J=6.9Hz,1H,10-H),6.68(d,J=10.0Hz,1H,4-H),5.46(d,J=10.1Hz,1H,3-H),5.06(s,2H,32,37-H),3.79(m,4H,morpholino-H),3,44(m,2H,11-H),3.30(s,2H,31-H),2.54(s,2H,22,26-H),2.31(m,4H,morpholino-H),2.12(m,2H,21-H),1.90~1.81(m,4H,piperidine-H),2.06(s,2H,21,36-H),1.75(s,3H,25-H),1.73(m,3H,29-H),1.69(s,3H,20-H),1.66(m,6H,34,39-H),1.60(s,3H,35-H),1.57(s,3H,40-H),1.45(2,3H,24-H),1.38~1.33(m,4H,paperidine-H);MSm/z: 780.5{[M+H]+};HR-MSm/z:Calcd for C47H60N2O8[M]780.4344,found 780.4343。
6e:产率53%;1H NMRδ: 11.90(s,1H,6-OH),7.45(d,J=7.2Hz,1H,10-H),6.66(d,J=10.4Hz,1H,4-H),5.46(t,J=7.9Hz,1H,3-H),5.07(s,2H,32,37-H),3,49(m,2H,11-H),3.30(m,2H,31-H),2.52(s,2H,22,26-H),2.32(m,2H,21-H ),2.08(m,2H,36-H),1.90~1.81(d,J=7.2Hz,4H,piperidine-H),1.75(s,3H,25-H),1.71(m,2H,20-H),1.67(s,6H,29,34-H),1.67(m,6H,34,29-H),1.57(s,9H,39,35,40-H),1.53(s,3H,24-H),1.43~1.28(m,12H,piperidine-H),1.12(m,4H,19-H);MSm/z: 779.5{[M+H]+}。
6f: 产率47%;1H NMRδ: 11.91(s,1H,6-OH),6.64(d,J=12.1Hz,1H,4-H),5.96(d,J=9.1Hz,1H,27-H),5.40(m,1H,3-H),5.05(s,2H,32,37-H),3.66~3.59(m,8H,2′,6′,2″,6″-H),3.28(m,2H,31-H),3.19(s,1H,11-H),3.15(s,1H,26-H),2.76(m,4H,3′,5′-H),2.54(m,2H,22-H),2.44(m,4H,3″,5″-H),2.08(m,4H,21,36-H),1.85~1.80(m,3H,25-H),1.75(s,3H,29-H),1.66(s,3H,20-H),1.56(s,6H,34-H),1.40(s,6H,39-H),1.37(s,3H,35-H),1.32~1.30(m,6H,40,24-H),1.09(s,3H,19-H);MSm/z: 785.5{[M+H]+};HR-MSm/z:Calcd for C46H60N2O9[M]784.4296,found 784.4293。
1.2 体外抗肿瘤活性测定
取培养的对数生长期贴壁肿瘤细胞: RKO、 HepG-2和OVCAR-3各一瓶,经胰酶消化液消化处理后用DMEN高糖培养基(含10%胎牛血清)调至约105个·mL-1(台盼蓝染色,普通光学显微镜下活细胞计数);在96孔培养板上每孔加入100μL(约含1.0×104个肿瘤细胞)培养液,置于含5%CO2,100%湿度的37℃恒温箱中培养24h。待测不同浓度化合物用DMEN不完全培养基(不含胎牛血清)溶解,微滤除菌(0.22μm)后用DMEN不完全培养基分别稀释至浓度为10μmol·L-1,5μmol·L-1,2.5μmol·L-1,1μmol·L-1和0.5μmol·L-1;每组设5个平行孔,以100μL不完全培养基作空白对照组。将96孔板在培养箱中温育24h后每孔加入MTT(溶于不完全培养基)10μL,再温育4h,吸出上清液,加入DMSO 150μL,振摇,用酶标仪于570nm处测定各孔光吸收差值,计算IC50值。
2 结果与讨论
2.1 合成
1与氢溴酸溶液反应的主要产物为2,而非预期的33-溴代转位藤黄酸。其可能原因是1的32,33-双键在HBr催化作用下与水发生了亲电加成反应,反应符合马氏规则,羟基连在33-位上得到2。由2的HR-MS分析可见,其分子量比1多18.01,说明1与一分子水发生了加成反应;由2的1H NMR分析可见,δ7.52出现10-H的吸收峰,而在δ5.10~5.00则未出现32,37-烯烃质子吸收峰,说明加成反应不是发生在9,10-双键,而是在32,33-或37,38-双键上;此外,2的1H NMR谱图中未出现1的3-H和4-H互相偶合的烯烃质子双峰,而出现了末端双键的信号,同时2比1少一个烯烃质子信号,由此推测2应为33-羟基转位藤黄酸。
合成3和4时,先用3-氨基-1-丙醇与相应的酰氯发生酰化反应合成中间体A和B,然后在碱碳酸铯催化下与1反应生成3和4。
合成6a~6f时,使用了DCC/HOSU反应体系。该反应的缺点是反应结束后会残留大量副产物N,N′-二环己基脲(DCU),我们利用DCU难溶于二氯甲烷的性质,将粗产物反复用二氯甲烷溶解、过滤后除去DCU。用1-OSU活化酯中间体分别与各种有机胺反应制备1的酰胺衍生物,比用1-(3-二甲氨基丙基)-3-乙基碳二亚胺(EDC)/DMAP体系,反应效率更高。
合成6f时,除在30-形成酰胺外,9,10-同时发生了迈克尔加成,在10-上连上了吗啡啉基。这一结果在6f的1H NMR和MS分析中得到验证:δ7.40~7.60未出现10-H烯烃质子信号,表明9,10-之间不再是碳碳双键,已经发生了加成反应;MS实验值(784.4293)与理论值(784.4296)非常接近。
2.2 体外抗肿瘤活性
2~6的抗肿瘤活性结果见表1。
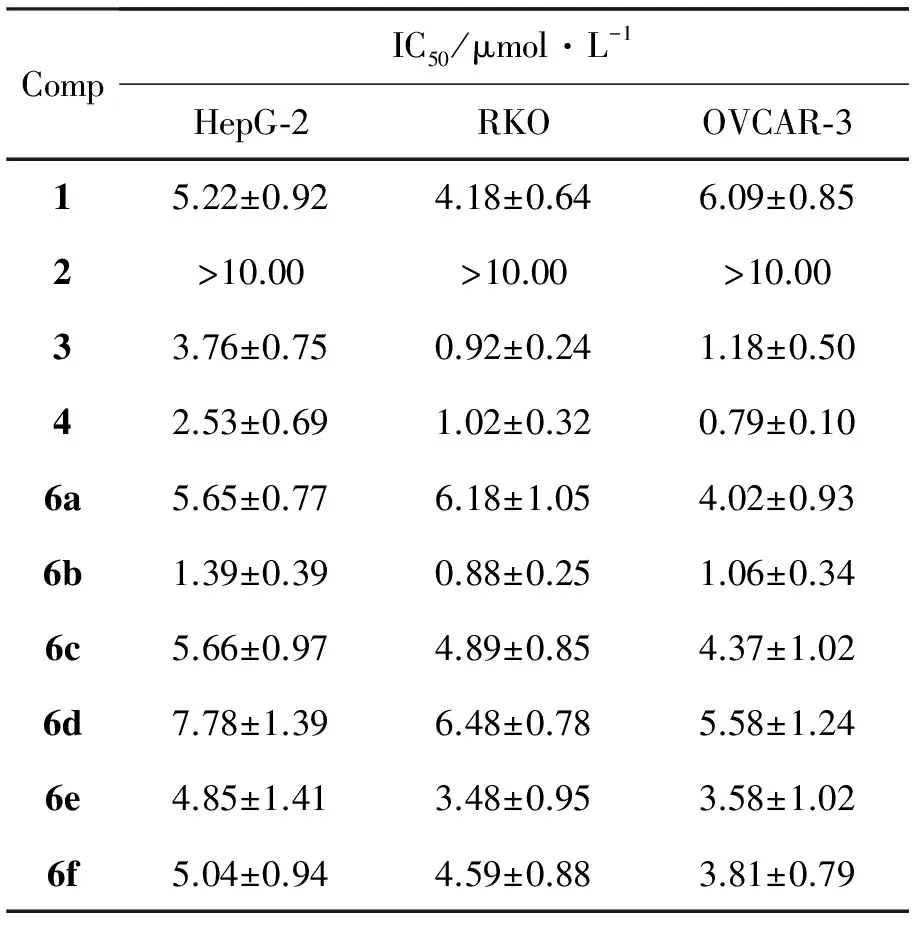
表 1 2~6对三种肿瘤细胞的IC50值Table 1 IC50 of 2~6 against HepG-2,RKO and OVCAR-3
由表1可见: (1)2对RKO,HepG-2和OVCAR-3的抗肿瘤活性大大低于1,说明在32,33-双键上发生加成反应,33-接上羟基后会导致抗肿瘤活性降低;但Wang J X等[16]发现33-氯代转位藤黄酸对人肺癌细胞(A549)、结肠癌细胞(HT-29)和人胃癌细胞(BGC-823)的抗肿瘤活性高于1,而对人肝癌细胞(Bel7402)的抗肿瘤活性却大大降低。由此可见,33-接上不同基团对1的抗肿瘤活性有不同的影响,接上亲脂性基团(如卤素等)比接上亲水性基团(如羟基等)有利;(2)6a,6c,6d,6e和6f对HepG-2和RKO的抗肿瘤活性与1相似。值得注意的是,6f的9,10-之间不是碳碳双键而是单键,但抗肿瘤活性仍高于1,这说明9,10-双键并不是1类似物诱导细胞凋亡所必需的。而此前报道[15-16]的实验结果显示,1的9,10-无论发生加氢还原反应、双羟基化反应或迈克尔加成反应在10-接上环己基,都会失去诱导细胞凋亡活性。由此可见,9,10-由双键变为单键后是否还会保留抗肿瘤活性,可能与10-连接的基团种类有关;(3)3,4和6b对RKO和OVCAR-3的抗肿瘤活性显著强于1,大约是其5~7倍。相对于6a,6c,6d和6e来说,3,4和6b的30-位引入的功能基团之间不是直接而是通过一短碳链连接,有利于其抗肿瘤活性。
3 结论
合成了9个新型的藤黄酸衍生物,并研究了其抗肿瘤活性。构效关系研究结果对今后工作有一定的指导意义。
[1] 王洋,张雪,马力艳,等.新型藤黄酸衍生物的合成[J].合成化学,2014,22(2):222-225
[2] 刘举,王洋,周云鹏,等.藤黄酸衍生物的合成[J].合成化学,2013,21(2):217-219
[3] 陈葆仁.藤黄抗癌成分的研究[J].江西医学院学报,1980,2:1-7
[4] 侯文洁,萧伟.藤黄酸的研究进展[J].中草药,2011,42(3):617-620
[5] 柳文媛,冯 锋,尤启冬.RP-HPLC法测定总藤黄酸中藤黄酸的含量[J].中草药,2003,34(8):706-707
[6] Guo Q L,Zhao L,You Q D,etal.Gambogic acid inducing apoptosis in humna gastirc adenocarinom SGC-7901cells[J].Chin J Nat Med,2004,2(2):106-110
[7] Wang Y,Chen Y,Chen Z,etal.Gambogic acid Induces death inducer-obliterator 1-meadiated apoptosis in Jurkat T cells[J].Acta Pharmacal Sin,2008,29(3):349-354
[8] Kasibhatla S,Jessen K A,Maliartchouk S,etal.A role for transferrin receptor in triggering apoptosis when trageted with gambogic acid[J].Proc Natl Acad Sci USA,2005,102(34):12095-12100
[9] Zhao W,Zhou S F,Zhang Z P,etal.Gambogic acid inhibits the growth apoptosis and cell cycle arrest[J].Oncol Rep,2011,25(5):1289-1295
[10] 张生烈,李乾,张磊,等.藤黄酸衍生物的合成及抗肿瘤活性研究[J].有机化学,2012,32:1450
[11] Zhang X J,Li X,Yang Y R,etal.Studies on chemical-structure modification and structure activity relationship of gambogic acid derivatives at carbon(34)[J].Chem Biodivers,2012,9(10):2295-2308
[12] Li,X,Zhang X J,Sun H P,etal.Synthesis and anti-tumor evaluation of novel C-37modified derivatives of gambogic acid[J].Chinese Journal of Chemistry,2012,30(5):1083
[13] Sun H P,Liu Z L,Xue X,etal.Studies on chemical structure modification and structure activity relationship of derivatives of gambogic acid at C(39)[J].Chem Biodivers,2012,9(8):1579
[14] He L Q,Ling Y,Fu L,etal.Synthesis and biological evaluation of novel derivatives of gambogic acid as anti-hepatocellular carcinoma agents[J].Bioorg Med Chem Lett,22:289-292
[15] Zhang H Z,Kasibhatla S,Wang Y,etal.Discovery,characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay[J].Bioorg Med Chem,2004,12:309
[16] Wang J X,Ma J H,You Q D,etal.Studies on chemical modification and biology of a natural product,gambogic acid(II):Synthesis and bioevaluation of gambogellic acid and its derivatives from gambogic acid as antitumor agents[J].European Journal of Medicinal Chemistry,2010,45(9):4343-4353.
SynthesisandAntitumorActivitiesofNovelGambogicAcidDerivatives
LI Jin-hai1a,1b,HUANG Yan1a,TAN Yin1a, ZHANG ZHI-jia1a,TAO Yi-wen1a,LIAO Si-yan1a
(a.School of Pharmaceutical Science;b.The Third Affiliated Hospital,1.Guangzhou Medical University,Guangzhou 510182,China)
Nine novel Gambogic acid derivatives (2~6)were synthesized by the reaction from Gambogic acid(1)with hydrobromic acid and organic amine,respectively.The structures were characterized by1H NMR,MS and HR-MS.Theinvitroantitumor activities of2~6against RKO,HepG-2and OVCAR-3were investigated by the MTT method.The results showed that (N-propyl-P-toluenesulfonamide)Gambogate(3),(N-propylphenylalaninamide)Gambogate(4)andN-tryptamine Gambogamide(6b)exhibited better antitumor activities than1,while the activity of 30-hydroxygambogellic acid(2)remarkablely declined.
gambogic acid;derivative;synthesis;antitumor activity
2013-09-30;
2014-09-23
广东省自然科学基金资助项目(S2011040000131);广东省科技计划资助项目(20BB031800021)
黎金海(1988-),男,汉族,广东肇庆人,硕士研究生,主要从事药物化学的研究。E-mail: jinyulao110@163.com
黄雁,博士,教授,E-mail: drhuangyan@163.com;谭茵,高级实验师,E-mail: shwhxsy@gzhmc.edu.cn
O623.624;O623.626
A
1005-1511(2014)06-0753-06
猜你喜欢
浙江化工(2024年2期)2024-03-15 02:27:40
健康体检与管理(2022年2期)2022-04-15 22:33:17
油气·石油与天然气科学(2021年12期)2021-12-11 01:43:23
洛阳理工学院学报(自然科学版)(2020年1期)2020-05-15 09:24:02
临床医药文献杂志(电子版)(2020年23期)2020-02-28 03:59:52
分析化学(2017年12期)2017-12-25 12:43:03
合成化学(2015年1期)2016-01-17 08:53:55
吉林大学学报(医学版)(2015年3期)2015-12-17 07:47:42
杭州师范大学学报(自然科学版)(2015年5期)2015-03-20 01:13:42
安徽中医药大学学报(2014年2期)2014-06-19 13:22:16
