
AFI型分子筛吸附H2分子的密度泛函理论研究
2014-07-19 07:58吴玉花苏暐光马晓琴赵天生冀永强白红存
石油学报(石油加工) 2014年1期
吴玉花,苏暐光,马晓琴,赵天生,冀永强,白红存,梁 彬
(1.宁夏大学 化学化工学院 省部共建天然气转化国家重点实验室培育基地,宁夏 银川750021;2.宁夏工商职业技术学院 化工系,宁夏 银川750001)
H2是重要的清洁能源。随着科学技术的发展,H2被广泛应用于光导纤维通讯、氢能源汽车、太阳能电池、石油化工、医药等方面[1-5];在电真空器件、半导体器件和集成电路的生产过程中,高纯度H2环境也异常重要[6-7]。近年来,氢能技术的研究取得了很大进展,但尚未成熟,其中H2的储存是使用氢能必须克服的难题之一。目前,储氢方法有气态储存、液氢储存、金属氢化物储存、有机物储存、多孔吸附剂上的吸附储存等[4,8-9]。其中,吸附储存是很具竞争力的储氢方法,而吸附材料则是关键。
分子筛由于具有较高的吸附量和选择性成为H2吸附贮存的重要吸附剂材料[10-11]。研究 H2分子在分子筛吸附剂上的吸附,从而获得相关基础数据,认识分子筛对于H2吸附的相位和吸附作用本质,非常必要和有意义。另一方面,随着量子化学方法的完善以及计算机软硬件的发展,量子化学计算已经广泛用于物质和分子的电子结构、功能性质和复合体系相互作用本质的研究[12]。对H2分子在分子筛孔道内的吸附进行理论计算和探讨同样具有高度的重要性。
在本研究中,笔者基于密度泛函理论考察了AFI分子筛H2分子的吸附,探讨了6种吸附模式下分子筛吸附H2分子的相互作用本质,以期对分子筛的H2吸附特性有更全面和深入的认识。
1 AFI型分子筛吸附H2分子的模型与计算方法
AFI是重要的具有直孔道结构的分子筛,其空间群为P6/mcc,属六方晶系,晶胞参数a、b、c、α、β、γ分别为1.38270nm、1.38270nm、0.85800nm、90.0°、90.0°、120.0°[13]。AFI分子筛[001]方向的孔道尺寸为0.73nm×0.73nm,笔者将H2分子置于该孔道中,研究其吸附行为。计算中,选择由36个Si原子和60个O原子为骨架的有限长分子筛孔道作为模型,其边界加上H原子,使模型的悬垂键得到饱和,如图1所示。
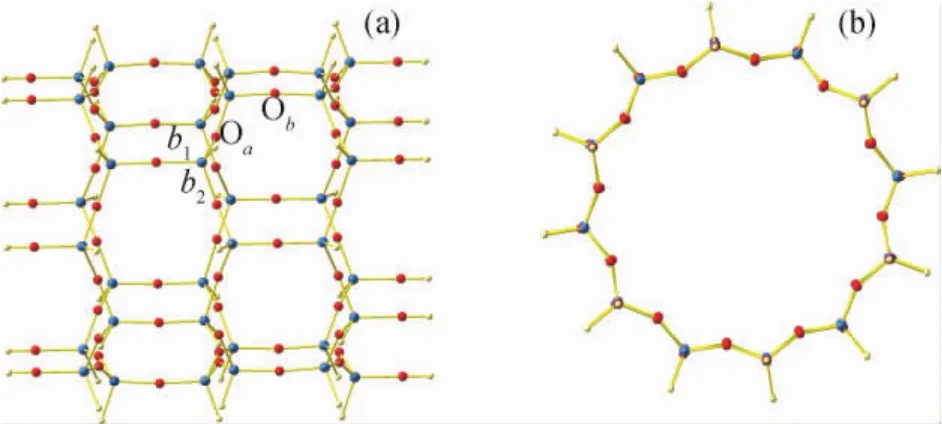
图1 计算中采用的AFI分子筛的模型Fig.1 The AFI zeolite model adopted in the calculations
笔者探讨的AFI型分子筛吸附H2分子的6种不同吸附模式如图2所示。图2中,S1表示H2分子垂直吸附Si原子,S2表示H2分子垂直吸附第1类O原子Oa,S3表示H2分子垂直吸附第2类O原子Ob,S4表示H2分子平行吸附平行Si—O键b1,S5表示H2分子平行吸附斜向Si—O键b2,S6表示H2分子沿键轴方向吸附在分子筛孔道中心。
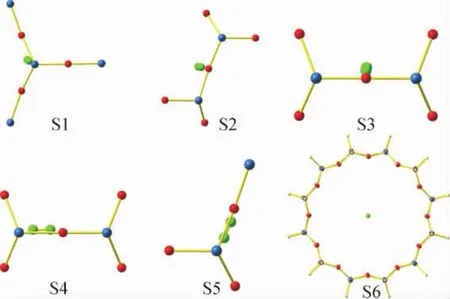
图2 H2分子在AFI分子筛中6种吸附模式(S1~S6)Fig.2 Six adsorption models(S1-S6)of H2 molecule in AFI zeolite
对上述所设计的AFI分子筛吸附H2分子模型,采用自洽场分子轨道法(Self-consistent field molecular orbital,SCF-MO)在 密 度 泛 函 理 论(Density functional theory,DFT)框架下进行计算。对于DFT的交换和相关函数,采用了Perdew、Burke和Ernzerh提出的PBE方法[14-15]。该方法已经成功地用于探讨多种小分子在限域空间内的吸附作用[16-19]。对于能量、电子性质、Mulliken布居分析的计算均采用密度泛函理论中的PBE方法,在高斯型基函数6-31G*水平上进行。进行能量和电子性质计算前,对所有模型进行了部分几何结构优化,即对吸附复合物模型只优化H2分子和吸附H2分子附近相关的O—Si—O或Si—O—Si单元,其他原子则固定在其晶体位置。这样的优化过程既保证了可以对分子筛的吸附位附近原子和吸附质分子在一定程度上进行结构调整,又使整个分子筛保持原有AFI骨架结构。使用相同方法在3-21G基组水平上对几何构型优化及振动分析计算。执行本研究的所有计算均使用Gaussian 09程序包[20]。
2 结果与讨论
2.1 AFI型分子筛吸附H2分子模型的几何结构
几何构型优化后获得的AFI型分子筛吸附H2分子的模型结构如图3所示,相应的键长和距离参数列于表1。由图3可见,几何构型优化后,除S3吸附模式外,其他5种吸附模式都基本保持了原有的吸附方式,只是相应的原子位置和相对距离有一定的调整。对于S3而言,优化前后发生了较大的变化。几何构型优化前,S3为H2分子垂直吸附第2类O原子Ob的构象;几何构型优化后,演变为H2分子平行吸附Si—O键b1的模式,即与S4中吸附模式近乎相同。因此,H2分子以垂直相位吸附于O原子时,其可能的构象是垂直吸附Oa类O原子,而不是垂直吸附Ob类O原子。
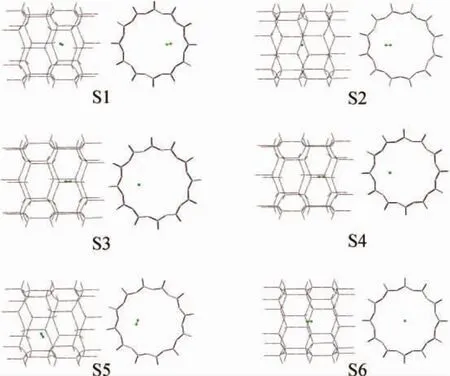
图3 经构型优化计算获得的H2分子在分子筛中吸附的结构Fig.3 Structures of H2adsorption in AFI zeolite obtained by geometrical optimizations
由表1可见,H2分子与分子筛骨架中最邻近Si原子的距离dSi…H均大于0.33nm,与最邻近O原子的距离dO…H均大于0.25nm。这些距离都远大于化学键作用范围,因此6种吸附方式均属于物理吸附范畴。在S1~S5中,dSi…H在0.3353~0.3581nm范围,dO…H在0.2549~0.3410nm范围。H 原子的范德华半径约为0.12nm,Si原子和O原子的范德华半 径 分 别 约 为 0.22 和 0.15nm[21-22]。 可 见,S1~S5中H原子与最邻近Si原子的相互作用都在范德华作用范围附近,而H原子与最邻近O原子的相互作用在范德华作用范围附近的只有S1和S2。此外,由于H2分子在S6中仍处于AFI分子筛孔道的管轴上,H原子与最邻近Si原子、O原子的距离都在0.5nm以上,明显大于范德华作用范围。由此可推断,S6中H2分子与分子筛骨架原子的相互作用将非常弱。
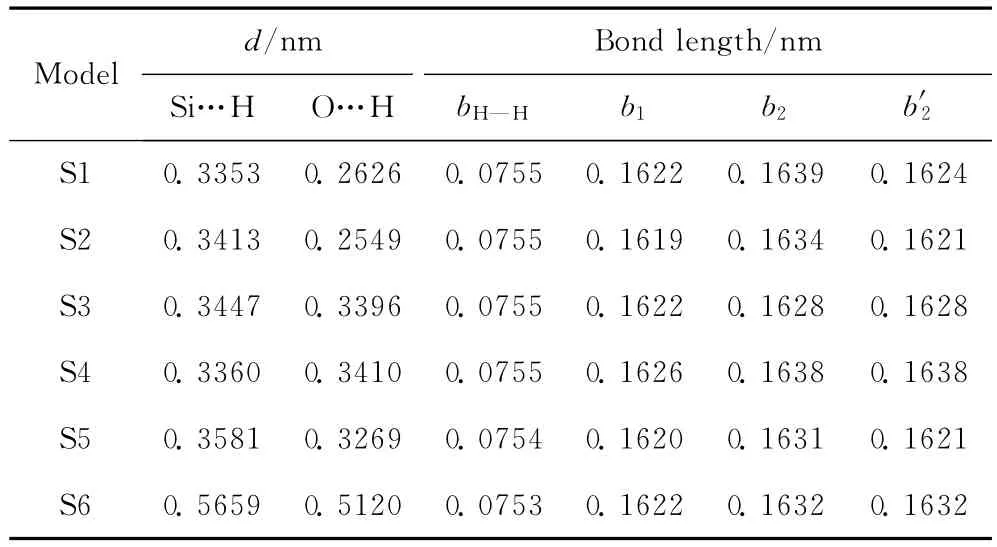
表1 AFI分子筛吸附H2分子模型S1~S6模式中的键长及相关距离参数Table 1 The bond lengths and the distances of S1—S6 models for H2adsorption in AFI zeolite
计算获得的S1~S6中H2分子的键长bH—H在0.0753~0.0755nm范围,与单独的 H2分子的键长0.0754nm相比,差别仅为±0.0001nm。由此可见,外围分子筛骨架对于H2分子的键长几乎没有影响。未吸附H2分子前,AFI分子筛骨架中2种Si—O键b1和b2键长分别为0.1622和0.1631nm。在S1~S6中,b1在0.1619~0.1626nm范围,b2在0.1621~0.1639nm范围,吸附前后键长的变化均小于0.001nm。由于6种模式中,H2分子的吸附都属于物理吸附,相互作用较弱,对于分子筛骨架几何结构的影响很小。
2.2 AFI型分子筛吸附H2分子模型S1~S6模式的能量与相对稳定性
表2列出了PBE/6-31G*方法水平上计算得到的S1~S6模式的能量(Es)。从能量的角度考虑,体系的能量越低,其热力学稳定性越高。因此严格地从相对能量的角度出发,S4即H2分子平行吸附b1型Si—O键模式具有最高的热力学稳定性。在此需要指出的是,S1与S4在能量上非常接近,仅相差0.01eV,在密度泛函计算的误差之内[23],可以理解为S1与S4的热力学稳定性非常接近。尽管S3经过几何构型优化后也演变为H2分子平行吸附b1型Si—O键模式,但其能量仍比S4高0.084eV,即S3不如S4稳定。本研究所考虑的6种模式中,S6具有最高的能量,因此其稳定性最低,原因可能是S6中H2分子与分子筛骨架的Si原子和O原子距离都很远,作用非常弱。
为了考察H2分子吸附过程的能量变化,定义AFI分子筛吸附H2分子的结合能Eb如式(1)所示。Eb=Es-EAFI-EH2(1)式(1)中,Es为AFI分子筛吸附H2分子复合体系的能量,EAFI和EH2分别为组成复合体系的2个分子片段AFI分子筛和H2分子的能量。由于笔者的研究对象是AFI分子筛吸附H2分子形成的复合体系,存在基组重叠误差(Basis set superposition error,BSSE)效应[23],直接计算一般会过高估计实际体系的结合能。因此,将考虑BSSE效应的影响得到的经过BSSE校正的结合能(Eb-BSSE)也列于表2。
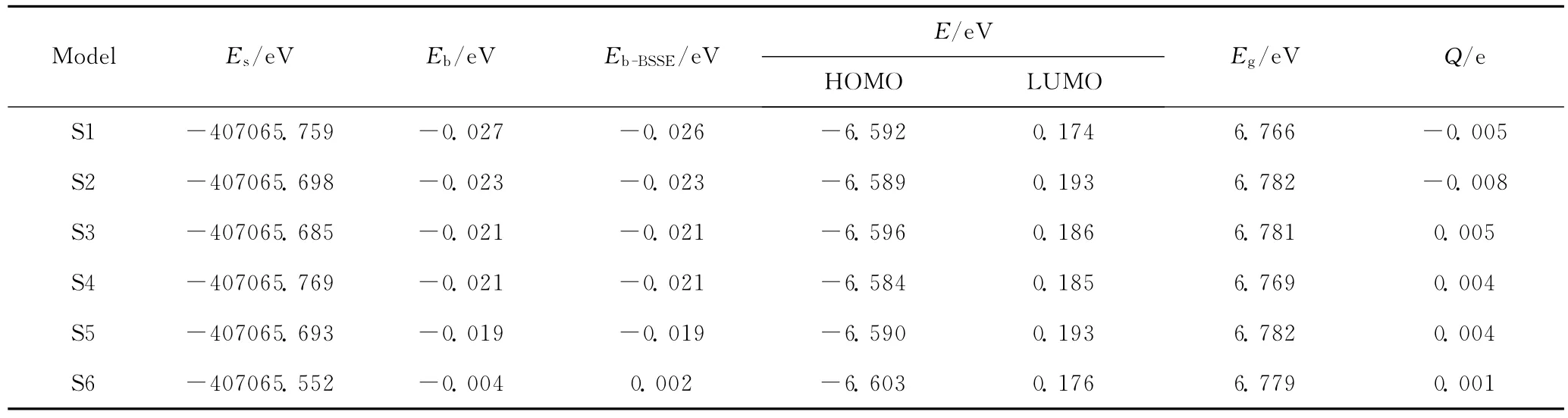
表2 分子筛吸附H2分子模型S1~S6模式的能量与电子性质Table 2 The energies and electronic properties of S1—S6models for H2adsorption in AFI zeolite
由表2可知,未考虑BSSE的结合能均为负值,即6种吸附模式下H2分子在AFI分子筛孔道内的吸附都是放热过程。其数值在-0.004~-0.027eV之间。S1放出的热量最多,而S6放热最少。陈媛梅等[24]使用相同方法计算表明,H2分子吸附于碳纳米管时,垂直吸附C原子和平行吸附C—C键2种模式的结合能分别为-0.058和-0.023eV。可见,分子筛吸附H2分子的结合能要比碳纳米管小一些。反之,吸附于AFI分子筛中的H2分子比吸附在碳纳米管的H2分子更容易脱附。
考虑BSSE效应对于S1~S5的结合能几乎没有影响,其数值除S1相差0.001eV外,其他均给出相同数值;但是S6的结合能变为正值,即H2分子在AFI分子筛孔道中央延轴向吸附为吸热过程。因此,不论是否考虑BSSE效应,S1均为最有利的吸附模式,S2次之,S3~S5再次之,S6最差。从结合能的角度出发,也印证了S6中2个分子片段间的相互作用非常弱。导致上述结合能大小次序的可能原因在于,S1和S2中,dSi…H约为0.34nm,dO…H约为0.26nm,H原子与分子筛骨架上的Si原子和O原子都处于范德华作用范围内;在S3~S5中,dSi…H在0.34~0.36nm 范围,dO…H大于0.33nm,只有Si原子与H原子处于范德华作用范围内,O原子与H原子不在范德华作用范围内;在S6中,H原子与Si原子和O原子距离均超过了范德华作用范围。此外,S3经几何构型优化后,演变为S4的H2分子平行吸附b1型Si—O键,不论是否考虑BSSE效应,二者的结合能数值均相同,这是由于S3与S4具有近乎相同的几何结构和吸附模式。因此,不同吸附模式导致的H2分子与分子筛骨架原子之间不同的范德华作用可能是影响其能量和热力学稳定性的主要因素。
2.3 AFI型分子筛吸附H2分子模型S1~S6模式的电子性质
分子的前线轨道尤其是最高占据轨道(The highest occupied molecular orbital,HOMO)、最低未占据轨道(The lowest unoccupied molecular orbital,LUMO)以及 HOMO-LUMO能级差(Eg)在化学反应中具有突出重要的作用[25]。计算了S1~S6的HOMO、LUMO以及Eg,结果也列于表2。由表2可知,AFI分子筛吸附H2分子复合体系的HOMO 能 级 在 -6.584 ~ -6.603eV 范 围,LUMO能级位于0.174~0.193eV范围,Eg范围在6.766~6.782eV;S1~S6电子性质的参数都非常接近,各体系HOMO、LUMO和Eg相差均在0.02eV以内。由此推断,不同吸附模式对于复合物电子性质的影响非常小。这主要源于H2分子吸附分子筛形成的复合物中,2个分子片段间属于范德华相互作用,轨道相互作用很弱。
对独立的2个分子片段AFI分子筛和H2分子也进行了计算,得到的AFI分子筛模型的HOMO、LUMO和Eg分别为-6.602、0.154和6.756eV,而单独H2分子的相应数值为-10.322、2.090和12.412eV。由此推断,AFI分子筛吸附H2分子形成的复合物的HOMO和LUMO应该都主要是原分子筛相应前线轨道的贡献,只是相应的HOMO和LUMO轨道能级高低有所变化。为了验证该推断,笔者绘制了AFI分子筛与H2分子吸附分子筛复合物的HOMO和LUMO空间分布图。由于S1~S6的轨道分布近乎相同,只列出了AFI分子筛和S1吸附模式下复合物的HOMO和LUMO空间分布图,如图4所示。由图4可见,AFI分子筛模型的HOMO主要来自分子筛骨架O原子的贡献,LUMO中骨架O原子和Si原子都有贡献;S1的HOMO和LUMO均来自分子筛骨架原子的贡献,H2分子没有贡献。S1的前线轨道空间分布与未吸附的体系相比,二者的差别非常小。这也验证了H2分子与分子筛骨架之间相互作用很微弱,分子片段间的轨道杂化现象几乎不存在[26]。
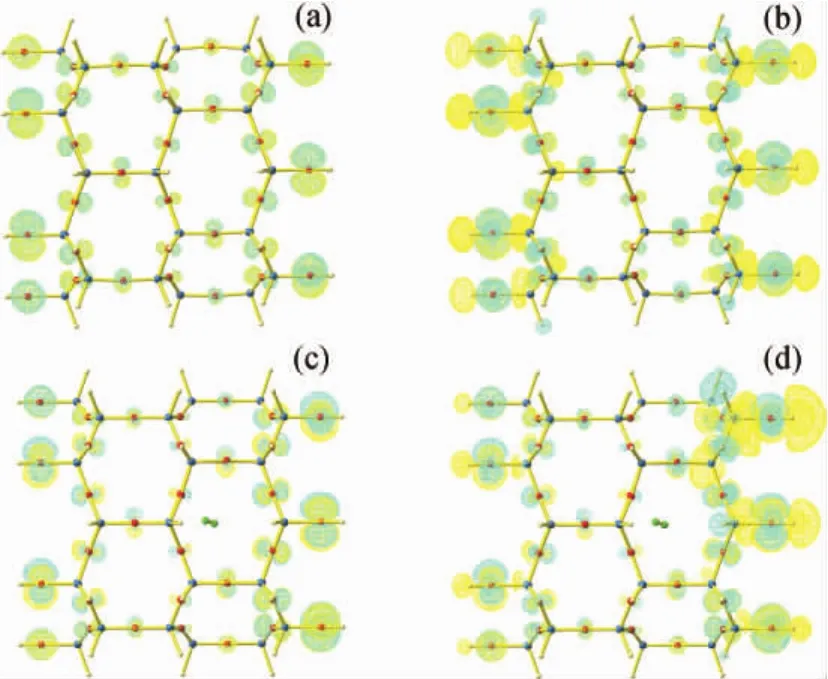
图4 AFI分子筛模型和S1模式的HOMO和LUMO空间分布Fig.4 The distributions of HOMO and LUMO for AFI zeolite and S1model
由表2还可知,S1和S2中,基于Mulliken电荷布居分析的电荷迁移Q为负值,即电子由分子筛骨架向吸附的H2分子迁移。S3~S6中则为电子由H2分子向分子筛骨架迁移。但是不论何种吸附模式,电荷迁移量都很少。最多的电荷迁移发生在S2中,但迁移到H2分子上的电荷也仅为0.008e;S6电荷迁移量尤其少,仅为0.001e。如此少的电荷迁移量对于H2分子以不同模式在分子筛中的吸附的影响也就很小。
3 结 论
(1)研究的AFI型分子筛吸附H2分子模型的6种吸附模式中,H2分子与分子筛之间相互作用均属于物理吸附范畴。由于相互作用较弱,吸附前后H2分子和AFI分子筛的结构变化都较小。H2分子以垂直相位吸附于O原子时,其可能的构象是垂直吸附Oa类O原子,而不是垂直吸附Ob类O原子。
(2)研究的AFI型分子筛吸附H2分子模型的6种吸附模式中,S4能量最低,S1与S4的能量非常接近;S1~S5中H2分子吸附均为放热过程,其中S1放热量最多;S6考虑BSSE效应时为吸热过程。
(3)AFI型分子筛吸附H2分子的不同吸附模式导致的不同范德华作用可能是影响其能量和热力学稳定性的主要因素,而吸附模式对于复合物电子性质的影响则很小。前线轨道分析表明,AFI分子筛吸附H2分子形成的复合物的HOMO和LUMO都主要由原分子筛相应前线轨道贡献,H2对分子前线轨道没有贡献。
(4)AFI型分子筛吸附H2分子模型的6种吸附模式中,S1和S2中的电子由分子筛骨架向吸附的H2分子迁移,其他4种吸附模式的电子由H2分子向分子筛骨架迁移,但电荷迁移量都很少。
[1]VILLA K,DOMENECH X, MALATO S,et al.Heterogeneous photocatalytic hydrogen generation in a solar pilot plant[J].Int J Hydrogen Energy,2013,38(29):12718-12724.
[2]DUNN S.Hydrogen futures:Toward a sustainable energy system?[J].Int J Hydrogen Energy,2002,27(3):235-264.
[3]HORIUCHI Y,TOYAO T,TAKEUCHI M,et al.Recent advances in visible-light-responsive photocatalysts for hydrogen production and solar energy conversion—from semiconducting TiO2to MOF/PCP photocatalysts[J]. Phys Chem Chem Phys, 2013, 15(32):13243-13253.
[4]CHALK S G,MILLERI J F.Key challenges and recent progress in batteries,fuel cells,and hydrogen storage for clean energy systems[J].J Power Sources,2006,159(1):73-80.
[5]ZHOU L S,WANG X W,XUE W N,et al.Beneficial effects of hydrogen-rich saline against spinal cord ischemia-reperfusion injury in rabbits [J].Brain Res,2013,1517:150-160.
[6]DERYCKE V,SOUKIASSIAN P G,AMY F,et al.Nanochemistry at the atomic scale revealed in hydrogeninduced semiconductor surface metallization[J].Nature Mater,2003,2(4):253-258.
[7]LYDING J W,HESS K,KIZILYALLI I C.Reduction of hot electron degradation in metal oxide semiconductor transistors by deuterium processing [J].App Phys Lett,1996,68(18):2526-2528.
[8]ZUTTEL A.Materials for hydrogen storage[J].Mater Today,2003,6(9):24-33.
[9]YANG J,GRZECH A,MULDER F M,et al.The hydrogen storage capacity of mono-substituted MOF-5 derivatives:An experimental and computational approach[J].Microporous Mesoporous Mater,2013,171:65-71.
[10]DU X,WU E.Modeling of monolayer adsorption of hydrogen on ZSM-5zeolite by lattice density functional theory[J].Surf Interface Anal,2013,45(9):1358-1362.
[11]DEEG K S,GUTIERREZ-SEVILLANO J J,BUENOPEREZ R,et al.Insights on the molecular mechanisms of hydrogen adsorption in zeolites[J].J Phys Chem C,2013,117(27):14374-14380.
[12]KOHANOFF J.Electronic Structure Calculations for Solids and Molecules:Theory and Computational Methods [M]. Cambridge: Cambridge University Press,2006:108-130.
[13]BAERLOCHER C,MCCUSKER L B,OLSON D H.Atlas of Zeolite Framework Types[M].5th edition,Amsterdam:Elsevier,2007:12-13.
[14]PERDEW J P, BURKE K, ERNZERHO M.Generalized gradient approximation made simple [J].Phys Rev Lett,1996,77(18):3865(1-4).
[15]PERDEW J P,BURKE K,ERNZERHO M.Errata[J].Phys Rev Lett,1997,78(7):1396(1).
[16]QIAO W,BAI H,ZHU Y,et al.Structure and electronic properties of the double-wall nanotubes constructed from SiO2nanotube encapsulated inside zigzag carbon nanotubes[J].J Phys:Condens Matter,2012,24(18):185302(1-8).
[17]WANG Y,HUANG Y,YANG B,et al.Crystal orbital study on carbon chains encapsulated in armchair carbon nanotubes with various diameters[J].Carbon,2008,46(2):276-284.
[18]SA N,WANG G,YIN B,et al.Theoretical study on non-covalent functionalization of armchair carbon nanotube by tetrathiafulvalene molecule [J].Phys E,2008,40(7):2396-2399.
[19]QIAO W,LI X,BAI H,et al.Structure and electronic properties of the nanopeapods-one dimensional C60O polymer encapsulated in single-walled carbon nanotubes[J].J Solid State Chem,2012,186:64-69.
[20]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09 [Z]. Wallingford CT: Gaussian,Inc,2009.
[21]胡盛志,周朝晖,蔡启瑞.晶体中原子的平均范德华半径 [J].物 理 化 学 学 报,2003,19(11):1073-1077.(HU Shengzhi,ZHOU Chaohui,CAI Qirui.Average van der Waals radii of atoms in crystals[J].Acta Phys-Chin Sin,2003,19(11):1073-1077.)
[22]BATSANOV S S.Van der Waals radii of elements[J].Inorg Mater,2001,37(9):871-885.
[23]FORESMAN J B,FRISCH E.Exploring Chemistry with Electronic Structure Method[M].Second Edition.Pittsburgh:Gaussian Inc,1996:98-99.
[24]陈媛梅,李卿,黄元河.H2分子在碱金属掺杂碳纳米管上的吸附特性[J].高等学校化学学报,2010,31(6):1235-1239.(CHEN Yuanmei,LI Qing, HUANG Yuanhe.Properties of H2adsorption on the alkali-metadoped carbon nanotube [J].Chem J Chinese Univ,2010,31(6):1235-1239.)
[25]AIHARA J. Weighted HOMO-LUMO energy separation as an index of kinetic stability for fullerenes[J].Theor Chem Acc,1999,102(1-6):134-138.
[26]ALBRIGHT T A,BURDETT J K,WHANGBO M.Orbital Interactions in Chemistry[M].Toronto:Wiley-Interscience,1985:18-21.structure of vanadium in Arabian heavy petroleum:An X-ray absorption investigation[C]//Int Acad Publ,1991:821-826.
[22]HOCKING M B,PREMOVIC P I.Coal inclusions of the Athabasca tar sands:Characterization and direct determination of vanadyl porphyrin content by electron spin resonance[J].Geochim Cosmochim Acta,1978,42:359-365.
[23]刘勇军,付庆涛,刘晨光.渣油加氢脱金属反应机理的研究进展[J].化工进展,2009,28(9):1546-1552.(LIU Yongjun, FU Qingtao, LIU Chenguang.Advances in reaction mechanism of residua hydrodemetallization [J]. Chemical Industry and Engineering Progress,2009,28(9):1546-1552.)
猜你喜欢
大学物理(2022年9期)2022-09-28
科学技术创新(2022年36期)2022-02-13
科学技术创新(2022年36期)2022-02-01
物理通报(2020年7期)2020-07-01
中国机械工程(2019年7期)2019-04-23
都市家教·上半月(2017年7期)2017-08-15
原子与分子物理学报(2015年3期)2015-11-24
石油化工应用(2014年12期)2014-03-11
石油化工应用(2014年2期)2014-03-11
郑州大学学报(工学版)(2014年6期)2014-03-01
