
基因组二代测序数据的自动化分析流程
2014-05-25 00:32:53李文轲李丰余张思瑶蔡斌郑娜聂宇周到赵倩
遗传 2014年6期
李文轲, 李丰余,, 张思瑶, 蔡斌, 郑娜, 聂宇, 周到, 赵倩
1. 中国医学科学院, 北京协和医学院, 国家心血管病中心, 阜外心血管病医院, 心血管疾病国家重点实验室, 北京 100037;
2. 中南民族大学生物医学工程学院, 武汉430074
基因组二代测序数据的自动化分析流程
李文轲1, 李丰余1,2, 张思瑶1, 蔡斌1, 郑娜1, 聂宇1, 周到2, 赵倩1
1. 中国医学科学院, 北京协和医学院, 国家心血管病中心, 阜外心血管病医院, 心血管疾病国家重点实验室, 北京 100037;
2. 中南民族大学生物医学工程学院, 武汉430074
二代测序技术的发展对测序数据的处理分析提出了很高的要求。目前二代测序数据分析软件很多, 但是绝大多数软件仅能完成单一的分析功能(例如:仅进行序列比对或变异读取或功能注释等), 如何能正确高效地选择整合这些软件已成为迫切需求。文章设计了一套基于perl语言和SGE资源管理的自动化处理流程来分析Illumina平台基因组测序数据。该流程以测序原始序列数据作为输入, 调用业界标准的数据处理软件(如:BWA, Samtools, GATK, ANNOVAR等), 最终生成带有相应功能注释、便于研究者进一步分析的变异位点列表。该流程通过自动化并行脚本控制流程的高效运行, 一站式输出分析结果和报告, 简化了数据分析过程中的人工操作,大大提高了运行效率。用户只需填写配置文件或使用图形界面输入即可完成全部操作。该工作为广大研究者分析二代测序数据提供了便利的途径。
二代测序; 自动化数据分析; 流程; 变异检测
二代测序技术(Next-generation sequencing)大幅度降低了测序的时间和成本, 使得大规模测序逐渐成为常规的实验室研究和临床检测手段。测序产生的数据量急剧增加, 如何高效地分析这些数据, 已成为迫切需要解决的问题。目前, 分析序列信息的生物信息学软件纷繁复杂, 但基本上每个软件只能完成单一的分析功能, 实现一个完整的分析流程则需要对众多软件进行整合, 而手动串联的效率往往不尽人意; 同时, 这些软件需要在Linux工作环境下以命令行运行, 要求用户具备较好的计算机背景;另外, 即便一些实验室完成了分析流程的构建, 他们往往不会公开许多细节, 新用户仍然要从头建起。本研究致力于构建经典的二代测序数据分析流程, 并实现各个环节的高效自动化管理和分析, 减轻研究者前期的工作负担, 促进相关领域进一步对基因组测序研究项目的顺利开展。
1 数据的获取和分析流程的构建
1.1 Illumina测序数据
本流程适用于 Illumina测序平台产出的双端(Paired ends)测序数据。Illumina测序技术采用边合成边测序(Sequencing by synthesis, SBS)的方法[1],早期的GA测序仪测序读长只有100个碱基, 随着技术的改进, 目前的读长已经增加到 150个碱基(Hiseq2500), 甚至更高的 250个碱基(Miseq)。测序读长不断增加, 测序通量也在不断上升。Illumina Hiseq2500是目前世界上通量最高的测序平台, 最多可以在大约10 d的时间内测定3000亿个碱基——即6~7个人类全基因组或60~80个人类全外显子组的序列测定。
Illumina平台以 FASTQ 格式[2]存储测序结果,这也是本流程的输入文件。FASTQ文件记录内容包括所测的碱基读段和质量, 其数据格式如图1所示。每条读段(reads)占四行:第一行和第三行为读段识别码, 包含测序仪 SN号、产生读段的巷道(lane)、该读段的编号等信息; 第二行为读段测到的碱基序列; 第四行为所测到碱基的质量分数, 每一个碱基都会对应一个质量分数。
1.2 数据处理流程及软件简介
目前测序数据处理软件很多, 我们综合考虑了适用性和效率, 整合出了一套标准的数据处理流程。具体来说, 获得FASTQ格式的原始测序数据后,需要对数据进行以下处理:(1)使用 BWA软件把这些短序列和参考基因组进行对比, 确定短序列在基因组上的位置, 把短序列组装成完整的人类参考基因组; (2)使用Samtools软件把这些短序列调整成按一定顺序(1~22, X, Y, 其他)排列的序列, 并进行数据格式的转换; (3)使用 Picard软件把测序产生的冗余信息和噪声去掉; (4)使用GATK寻找样本测序数据与参考基因组的差异, 列出这些差异点; (5)使用Annovar对这些变异位点进行功能注释, 得到一个易于理解的变异位点列表。处理流程图如图 2 所示。

图1 FASTQ格式示例

图2 处理流程图
1.2.1 读句比对软件BWA
BWA(Burrows-Wheeler Alignment tool)是基于Burrows-Wheeler变换(Burrows-Wheeler Transform)[3]的序列比对软件, 能高效地比对短序列和参考基因组, 找到短序列在参考基因组上的位置, 该软件最长支持至1 Mb的短序列比对。BWT方法通过B-W转换将基因组序列按一定规则压缩并建立索引, 再通过查找和回溯来定位读段, 在查找时可通过碱基替代来实现允许的错配。采用Burrows-Wheeler转换的代表软件是Bowtie和BWA。比对结果如图3所示:界面上方是测到的短序列, 下方是短序列所比对到的参考基因组。
1.2.2 SAM文件处理软件Samtools
读段定位到基因组后推荐采用 SAM(Sequence Alignment/Map)[4]格式或其二进制版本BAM格式来存储。二进制版本可大大节省存储空间, 但不能直接用普通文本编辑工具显示。
SAM 文件处理软件 Samtools可以很好的对SAM/BAM 格式数据进行操作, 因此, 本文使用它来进行数据格式转换和排序。
1.2.3 测序噪声去除和测序数据评价软件Picard
对组装好的全基因组数据, 需要将过度重复测到的数据进行剔除, 并且需要对数据质量进行评价, Picard软件可以很好地完成这两项工作[5]。
1.2.4 变异检测软件GATK
GATK 主要用于在测序数据中寻找变异[6], 包括单碱基变异(SNV)、短插入缺失(INDEL), 是当前业界用来寻找变异的主流软件。

图3 比对结果示例利用Broad Institute的IGV(Integrated Genomics Viewer)对数据进行可视化, 图4同。
变异指测序序列和参考序列的差异。如图4所示, 参考序列上的碱基是胸腺嘧啶(T), 而测序数据上的碱基是鸟嘌呤(G), 说明此处有一个 T→G 的突变。
1.2.5 变异注释软件ANNOVAR
ANNOVAR是一个用于高效注释变异的工具[7]。注释信息包括变异所在的染色体, 开始位置, 结束位置, 参考序列信息和观察到的序列信息的列表。一个变异经过ANNOVAR注释之后, 其功能一目了然, 便于进一步的生物学分析。
2 自动化实现
2.1 基于Perl语言的流程设计
本数据处理流程主要使用Perl编程语言实现对各个软件的高效串接和自动化操作[8]。一项计算任务正在进行时, Perl对它进行监控; 当计算完成, Perl去查看它的输出的计算结果, 并把结果作为下一个计算任务的输入, 往计算节点上投放新的计算任务。如此循环, 直到流程运行完毕。
同时, 由于每次运行的样本不同, 数据的输入输出位置也有差异。如果每处理一个新的样本, 就要对流程源码进行大量修改, 将不利于流程的使用。为此, 本流程定义了一个配置文件(config file)。通过配置文件可以指定:流程处理的样品名、数据输入输出路径、参考序列文件, 甚至流程中涉及到的软件的位置、软件的运行方式; 另外, 我们还提供了对流程中主要软件参数的修改, 以满足高级用户需求。每次进行一个新样本的分析, 不需要修改主程序代码, 只要为其创建一个配置文件, 主程序会自动读取配置文件, 生成相应的执行代码。
流程文件构成如图5所示。

图4 单碱基突变示例

图5 分析流程结构
2.2 基于资源管理软件(SGE)的并行设计
流程的运行环境是计算机集群, 其有别于普通PC机, 一般是由一台管理主机来协调许多计算主机来完成大型的计算任务。根据这样的硬件特点来设计流程, 需要考虑以下两个问题:(1)如何让众多计算机协同工作; (2)程序设计尽可能让计算任务并行,充分利用计算资源, 缩短计算时间。
SGE(Sun Grid Engine)是使用最广泛的分布式资源管理器(DRM)。SGE软件为用户提供了SGE系统提交要求计算的任务的方法, 动态分配工作负荷[9]。主节点接受用户提交的计算任务, 根据计算节点的负载情况, 动态决定把计算任务分配到哪个计算节点上进行, 使众多计算机协同工作。
通过分析各个软件的工作方式, 我们对中间多个步骤进行了并行设计, 配合 SGE, 对计算机资源进行高效调用, 从而大大缩短流程运行时间。流程的并行设计如图6所示。
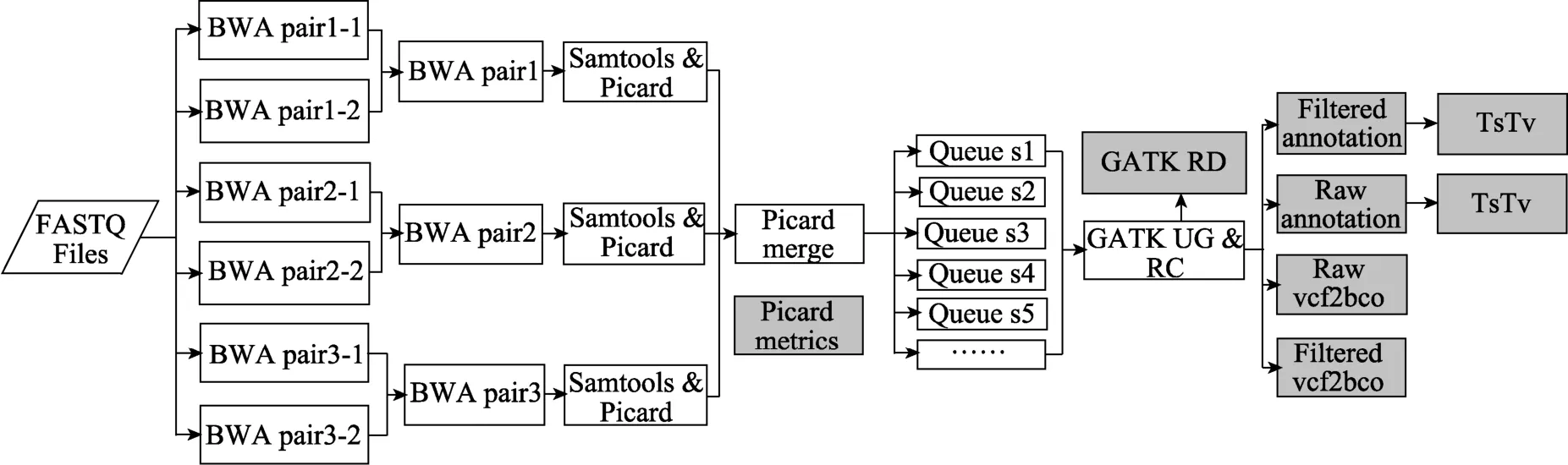
图6 流程并行设计
2.3 基于Java的图形界面设计
按照预定格式填写配置文件后, 本流程即可在终端直接运行, 不过为了进一步改善用户体验, 使操作更加更加简洁直观, 我们还提供了一个基于Java开发的图形界面:WGS_Pipeline_Runner。使用时, 用户可以直接调用已有的配置文件, 并进行修改, 也可以直接在表单界面进行填写, 完成后可以保存至本地。完成配置文件后, 点击 Run即可自动化完成分析流程, 一步输出分析报告。图形界面如图7所示。
本流程所涉及的所有软件说明、自动化代码及使用说明、配置文件说明等均可在 https://github.com/ wksofia/wgs_pipeline中下载使用。
3 流程测试
3.1 运行效率统计
一个135 GB的人类全基因组测序数据, 在计算机集群上使用该流程来处理大约耗时 50 h, 各阶段运行耗时如表 1所示。与该自动化流程相比, 在不考虑中间衔接耗时、不采用并行的情况下, 执行同样流程用时在一周以上。可见, 本流程不仅简化了分析操作, 更极大地节约了时间, 从而加速科研进展。
3.2 结果展示
自动化运行完全部流程, 得到一系列结果, 包括:BWA align读句定位生成的sai文件, BWA sampe整合pair-end信息得到的sam文件, Samtools convert转换sam得到的bam文件, Samtools sort对bam文件排序得到的sorted.bam文件, Picard rmdup去除重复得到的sample_duprmed.bam文件, GATK UG和GATK VQSR得到的一系列raw.vcf文件, Filter过滤后得到的 filtered.vcf文件, 以及 Annotation注释后的 csv变异文件。此外还给出了一个包含对实验数据质量评价的summary文件。综合以上结果, 用户能够从中挖掘出感兴趣的变异信息。
在这些结果中, 用户最值得关注的主要有两个文件:(1)经过功能注释的变异列表(见Annotation文件夹); (2)对实验数据质量的评价表(见sample.Summary)。
变异列表(部分)如表2所示。
每个个体大约会携带大约3百万个所谓的“变异”, 其中一些跟某些疾病的患病风险有关, 科研人员正是希望找到这种致病变异。表中每一行代表一个变异, 这个列表包含的信息主要有:这个变异在所在的基因, 变异的功能, 是否处于重复序列, 是否被前人报道过。
测序数据评价如表3所示。该表主要关注两个方面:平均测序深度和参考序列的覆盖度。

图7 图形界面

表1 流程详细运行时间
该评价表会在分析完成后, 通过电子邮件自动发送到用户邮箱, 既便于用户第一时间知道自己的数据质量, 也方便了监控。

表2 变异列表(部分)
4 结 语
本项目成功整合了一系列二代测序数据分析软件, 形成了一套经典的数据分析流程。本流程通过并行化设计和自动化处理, 一方面简化了操作成本、缩短了数据分析周期, 另一方面也使本流程可以引入更完善的数据校验步骤, 增强结果的可信度。本流程针对Illumina平台双端测序数据开发, 满足了大部分处理需求, 并对其他用户提供了一个很好的参考, 后续我们将根据用户需求对该自动化流程进行持续维护。

表3 测序数据评价表(部分)
随着二代测序技术的逐步发展, 二代测序已经广泛应用于科研和临床研究。本流程提高了二代测序数据分析的入门和运转效率, 其必将在二代测序相关基因组学研究中, 促进广大科研人员工作的高效进行。
[1] Illumina Inc. Illumina Sequencing Technology. http://www. illumina.com/documents/products/techspotlights/techspotl ight_sequencing.pdf.
[2] Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res, 2010, 38(6): 1767–1771.
[3] Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics, 2010, 26(5): 589–595.
[4] Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 2009, 25(16): 2078–2079.
[5] Picard. http://picard.sourceforge.net
[6] McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res, 2010, 20(9): 1297–1303.
[7] Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 2010, 38(16): e164.
[8] Scbwartz RL, Pboenix T, Foy BD著. 盛春, 蒋永清, 王晖译. Perl语言入门 (第五版). 南京: 东南大学出版社, 2009, 200.
[9] ORACLE INC. N1 Grid Engine 6 用 户 指 南 . http://docs.oracle.com/cd/E19080-01/n1.grid.eng6/817-7681/esqcr/index.html.
(责任编委:胡松年)
Automatic analysis pipeline of next-generation sequencing data
Wenke Li1, Fengyu Li1,2, Siyao Zhang1, Bin Cai1, Na Zheng1, Yu Nie1, Dao Zhou2, Qian Zhao1
1. State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Disease, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China;
2. College of Biomedical Engineering, South-Central University for Nationalities, Wuhan 430074, China
The development of next-generation sequencing has generated high demand for data processing and analysis. Although there are a lot of software for analyzing next-generation sequencing data, most of them are designed for one specific function (e.g., alignment, variant calling or annotation). Therefore, it is necessary to combine them together for data analysis and to generate interpretable results for biologists. This study designed a pipeline to process Illumina sequencing data based on Perl programming language and SGE system. The pipeline takes original sequence data (fastq format) as input, calls the standard data processing software (e.g., BWA, Samtools, GATK, and Annovar), and finally outputs a list of annotated variants that researchers can further analyze. The pipeline simplifies the manual operation and improves the efficiency byautomatization and parallel computation. Users can easily run the pipeline by editing the configuration file or clicking the graphical interface. Our work will facilitate the research projects using the sequencing technology.
next generation sequencing; automatic data analysis; pipeline; variantion detection
2013-09-07;
2014-01-20
国家重点基础研究发展计划(973计划)项目(编号:2010CB529505)和中央高校基本科研业务费专项资金(编号:2012-XHGX02)资助
李文轲, 硕士, 助理研究员, 研究方向:生物信息学。Tel: 010-88396071; E-mail: wksofia@gmail.com
赵倩, 博士, 副研究员, 研究方向:遗传学, 生物信息学。E-mail: zhaoqian82@gmail.com
10.3724/SP.J.1005.2014.0618
时间: 2014-3-25 17:22:30
URL: http://www.cnki.net/kcms/detail/11.1913.R.20140325.1722.002.html
猜你喜欢
电脑爱好者(2022年12期)2022-05-30 10:48:04
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
网络安全和信息化(2019年4期)2019-12-23 09:39:34
电脑爱好者(2019年20期)2019-12-10 06:30:31
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
网络安全和信息化(2018年5期)2018-11-09 02:02:26
