
建立携带左旋天门冬酰胺酶的红细胞特异性表达系统
2014-05-04 08:03潘欣曾思良宋维余秀珍张俊张鸿声方伟陈敏廖万清
生物技术通讯 2014年2期
潘欣,曾思良,宋维,余秀珍,张俊,张鸿声,方伟,陈敏,廖万清
1.上海俊维寓医馆,上海 200433;2.上海市中原中学,上海 200438;3.同济大学,上海 200433;4.第二军医大学,上海 200433
左旋天门冬酰胺酶(L-asparaginase,L-ASNase)是治疗淋巴细胞白血病等恶性血液病的重要药物之一[1]。某些类型的肿瘤细胞表达的L-天门冬酰胺合成酶(L-asparagines synthetase,L-AS)活性远远低于正常细胞,难以在胞内将氨或L-Glu的酰胺基在ATP和Mg2+存在时有效地转移至L-Asp的β羧基生成L-Asn,因而迫使这类肿瘤细胞利用外源Asn以维持胞内蛋白质合成与细胞增殖[2];而L-ASNase可水解Asn(必需氨基酸)为Asp和氨,由于人体内不存在天然L-ASNase,若向患者体内引入L-ASNase,全身性降解Asn,可使这类肿瘤细胞因L-AS活性低,不能独立合成Asn,造成Asn饥饿,导致蛋白质的生物合成紊乱而死亡,达到在不影响正常细胞生理功能的前提下杀伤肿瘤细胞的目的[3]。但L-ASNase是异源酶,免疫原性易致人体产生灭活酶的抗体,且不良反应较大,因此亟待研发低免疫原性L-ASNase给药方案。目前有临床试验将L-ASNase用急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)复发患者的自体红细胞(red blood cell,RBC)封装后再自体回输[4],该给药方案在理论上可消弱L-ASNase的抗原性,阻碍治疗性酶被宿主蛋白水解酶降解;在临床上也观察到可降低ALL复发患者抗-L-ASNase抗体的形成,明显减弱过敏反应、凝血功能障碍和肝功能失常,改善了病患的健康状况。然而,这种用非基因修饰方法将药物封装进RBC的方案或多或少均改变了RBC的形态和细胞膜特征(如易碎、变形性下降等),增加了机体清除这部分RBC的机率[5]。由于造血干细胞在体外向红细胞定向分化需时较长,若向造血干细胞中引入L-ASNase基因,可以使其在定向分化过程中开始表达L-ASNase,克服红细胞生命力短暂、难以在体外长期存活之弱点,延长药物时效,为寻找合适的分化期导入体内遏制ALL奠定基础。
在本研究中,我们用携带L-ASNase的红细胞特异性重组慢病毒载体系统包装出自灭活(self-inacti⁃vation,SIN)重组慢病毒,以小鼠红白血病(murine erythroleukemia,MEL)细胞经诱导可向红细胞终端分化为模型,探讨重组慢病毒感染后MEL细胞在分化过程中表达L-ASNase目的基因,延长药物时效的可行性。
1 材料与方法
1.1 材料
人胚胎肾(human embryonic kidney,HEK)293T细胞、MEL细胞、小鼠成纤维细胞NIH/3T3和稳定表达对照病毒RRL-sEF1α-GFPLuc的NIH/3T3细胞、人宫颈癌HeLa细胞为本课题组保存;大肠杆菌DH5α、包装质粒psPAX2、包膜蛋白质粒pMD2.G、转移质粒RS9-L-ASNase-2A-mCherry-PGK-MGMTHS4-IVS2(-)、RRL-sEF1α-L-ASNase-2A-mCherry和RRL-sEF1α-2A-mCherry为本课题组保存。
各种限制性内切酶为New England Biolabs公司产品;DMEM培养基、AIM-V XIVIVO-5培养基、胎牛血清(FBS)为Invitrogen Gibco公司产品,O6-苄基鸟嘌呤(O6-benzylguanine,BG)、双氯乙亚硝脲[1,3-bis(2-chloroethyl)-1-nitrosourea,BCNU;商品名为卡莫司汀]、六亚甲基二乙酰胺(N,N'-hexameth⁃ylene bisacetamide,HMBA)、聚乙烯亚胺(polyethyl⁃enimine,PEI)和聚凝胺(polybrene)为Sigma公司产品;兔抗大肠杆菌L-ASNase多抗为GeneTex公司产品;小鼠抗mCherry单克隆抗体为EarthOx公司产品;青霉素和链霉素溶液(以下简称双抗)为Hyclone公司产品;InstaGene Matrix为Bio-Rad公司产品;荧光定量(TaqMan)快速PCR混合试剂盒(2×)为ABI公司产品;哺乳动物细胞总蛋白抽提试剂(mammali⁃an protein extraction reagent,M-PER)、2,2-联喹啉-4,4-二甲酸二钠(bicinchoninic acid,BCA)蛋白定量分析试剂盒为Pierce公司产品;辣根过氧化物酶标记的山羊抗兔IgG和键合了Alexa 488的山羊抗兔IgG为Affinity Biologicals公司产品;标准品LASNase为日本协和发酵麒麟株式会社产品。
1.2 慢病毒转移载体的设计和制备
RS9-L-ASNase-2A-mCherry-PGK-MGMTHS4-IVS2(-)的结构(图1)中,为方便监测目的基因的表达,将编码L-ASNase的cDNA通过2A联接体技术与编码红色单体荧光蛋白mCherry的cDNA连接,同受β-珠蛋白启动子(promoter,P)、β-珠蛋白3'端增强子(enhancer,E)和β-珠蛋白位点控制区(locus control regions,LCR)DNaseⅠ高敏位点(hypersensi⁃tive site,HS)中具有较强增强子活性的HS2和具有染色质开放活性的HS3调控[6]。2A为源于一点褐翅蛾病毒的2A短肽(Thosea asigna virus 2A,T2A),当核糖体快速处理2A肽C端甘氨酰-脯氨酰之间的肽键合成时,会引起2A肽与其下游肽之间发生自裂解,该序列允许在同一读框中通过2A的自裂解表达多种非融合的独立的目的蛋白[7]。位于载体3'端可以非组织特异性地表达甲基鸟嘌呤甲基转移酶变异体(methylguanine methyltransferase,MGMT)P140K的耐药基因由人磷酸甘油酸激酶启动子(phospho⁃glycerate kinase promoter,PGK-P)控制,这样可以通过使用化疗药物BG和BCNU富集基因修饰的阳性细胞,并确保在筛选富集细胞的过程中目的基因受到有效控制,不会表达。为提高载体制备的有效性,增加慢病毒滴度和转染率,删除了慢病毒载体LCR区域中功能待定的HS1和有增强子活性的HS4,同时删除了β-珠蛋白中具内含子增强子活性的第 2个内含子(intervening sequence 2,IVS2)[8]。由于LCR主要的功能是激活β-珠蛋白基因簇,限制珠蛋白基因仅在红系细胞中表达,确保受调控基因在发育或分化过程中以一定的时序和模式正确表达,因而赋予该载体具有红系组织特异性[9]。
为快速测定慢病毒载体所携带的目的基因可否在靶细胞中表达,我们还构建了非组织特异性慢病毒载体 RRL-sEF1α-L-ASNase-2A-mCherry,利用2A联体技术将L-ASNase基因和mCherry报告基因置于短的延长因子 1α(short elongation factor 1α,sEF1α;删除了第一内含子)启动子控制下,在联体基因的3'端增加了土拨鼠肝炎病毒转录后调控元件(woodchuck hepatitis virus post-transcriptional reg⁃ulatory element),以提升mRNA的polyA加尾效率。为验证2A是否影响下游报告基因的正常表达,还构建了仅表达2A-mCherry的非组织特异性慢病毒载体RRL-sEF1α-2A-mCherry。
应用不同的限制性内切酶及DNA重组技术构建上述慢病毒转移载体。
1.3 重组慢病毒包装
用三质粒系统瞬时转染HEK 293T细胞制备重组慢病毒颗粒[9]。当HEK 293T细胞在10 cm细胞培养皿中生长达80%~90%汇合时,弃含双抗(青霉素终浓度为50 U/mL,链霉素终浓度为50 mg/mL)的完全DMEM培养基(含10%FBS),将PEI转染三质粒混悬液(质粒总量为8 μg/皿,pMD2.G、psPAX2和转移质粒按1∶2∶3的质量比例混悬于1 mL无血清无抗生素 DMEM 中,再加 24 μL pH7.0 的 1 μg/μL PEI)加至用9 mL含10%FBS的完全培养基覆盖的HEK 293T细胞中,24 h后弃上清,换预热的新鲜完全培养基,分别于48和96 h收获细胞培养上清中的病毒液,500 r/min离心10 min除去脱落的细胞和大的细胞碎片,用0.45 μm的滤器过滤上清,滤液经8500 r/min离心12 h浓缩病毒颗粒,用无血清的AIM-V XIVIVO-5培养基重悬病毒颗粒,分装至冻存管,-80℃保存备用。将重组慢病毒以载体名称命名。
1.4 重组慢病毒滴度测定
用终质量浓度为8 μg/mL的Polybrene介导重组病毒颗粒转染小鼠成纤维细胞NIH/3T3,培养7 d后用InstaGene Matrix抽提基因组DNA。以稳定表达对照病毒RRL-sEF1α-GFPLuc的NIH/3T3细胞基因组为对照,2套引物-探针联合使用,采用多重TaqMan PCR检测病毒滴度[9]。慢病毒序列检测所用正向引物为GAG-F(5'-GGAGCTAGAACGATTC GCAGTTA-3'),反向引物为 GAG-R(5'-GGTTGTA GCTGTCCCAGTATTTGTC-3'),探针为 GAG-P(5'-[FAM]ACAGCCTTCTGATGTTTCTAACAGGCCAGG[TAMRA]-3')。小鼠β-actin(简称BAC)为内源性对照,正向引物为BAC-F(5'-GGCACCACACCTTC TACAATG-3'),反向引物为 BAC-R(5'-GGGGTGT TGAAGGTCTAAAC-3'),探针为 BAC-P(5'-[HEX]TGTGGCCCCTGAGGAGCACCC[BHQ1]-3')。 用Applied Biosystems公司的7500 PCR仪,每个定量PCR反应中基因组DNA共使用100 ng(1×TaqMan快速PCR混合试剂,500 nmol/L基因特异性正、反向引物和探针),每组反应设3个重复。在SDS 2.1软件中设置定量PCR反应程序为95℃ 10 min热启动;PCR反应94℃ 15 s,60℃ 1 min,40个循环[10]。
已知稳定表达对照病毒RRL-sEF1α-GFPLuc的NIH/3T3每个细胞中有1个慢病毒载体拷贝数(vector copy number,VCN),其基因组DNA用未感染慢病毒的NIH/3T3细胞基因组DNA进行1/5系列稀释后,通过定量PCR获得慢病毒转染的VCN回归曲线。根据该曲线,计算未知样品中感染重组慢病毒GAG基因的拷贝数。病毒滴度(titer,T)计算公式:T(U/mL)=VCN×稀释率的倒数×起始细胞数[11]。
1.5 免疫染色
人宫颈癌HeLa细胞接种于6孔培养板的无菌盖玻片上,非组织特异性重组慢病毒RRL-sEF1α-L-ASNase-2A-mCherry按 1∶10稀释感染 HeLa细胞,24 h后换新鲜完全培养基,96 h后用PBS洗3次,用4%多聚甲醛固定30 min,PBS洗后用含0.5%Triton X-100的PBS透化30 min,用Image-iT FX信号增强剂封闭30 min,加入1∶500稀释的兔抗大肠杆菌L-ASNase多抗室温孵育2 h,PBS洗后,加入1∶1000稀释的键合了Alexa 488的山羊抗兔IgG,室温避光孵育60 min,PBS洗后,晾干的盖玻片用抗褪色金牌介质在载玻片上装片,指甲油封片。
1.6 BG/BCNU筛选与报告基因检测
为有效富集阳性转染细胞,MEL细胞经重组慢病毒感染后,先与50 μmol/L BG孵育1 h,再加50 μmol/L BCNU于37℃共孵育1 h,1 h后换预热的不含药物的新鲜培养基,以后每3 d换1次培养液,移除悬浮细胞,筛选14 d后进行后续实验。
分别在指定时间收获1×105经筛选富集的感染非组织特异性重组慢病毒的MEL细胞,用流式细胞仪检测mCherry红色荧光强度,以评估BCNU有效筛选情况。
用荧光显微镜监测感染组织特异性重组慢病毒的MEL细胞分别在5 mmol/L HMBA诱导前、后报告基因mCherry的表达情况,以评估调控因子对目的基因组织特异性表达的控制效果。
同时用流式细胞仪监测感染或未感染重组慢病毒的MEL细胞经5 mmol/L HMBA诱导前、后报告基因mCherry的表达情况,以评估MEL细胞向红系终端分化过程中携带目的蛋白的能力。
1.7 免疫印迹
在指定时间收获感染或未感染或诱导的MEL细胞,用哺乳动物细胞总蛋白抽提试剂抽提细胞总蛋白,用Bradford法进行蛋白定量,取等量总蛋白(每孔60 μg/20 μL)进行12%SDS-PAGE,然后转移至PVDF膜,室温下用50 g/L脱脂奶粉封闭1 h,膜与1∶1000稀释的一抗于4℃孵育过夜,洗膜后与HRP标记的二抗于37℃温育1 h,ECL显色,获取图像。检测L-ASNase印迹膜使用的一抗为兔抗大肠杆菌L-ASNase多克隆抗体,二抗为山羊抗兔IgG;检测mCherry的印迹膜使用的一抗为鼠抗mCherry单克隆抗体,二抗为兔抗鼠IgG;检测看家基因GAPDH表达的印迹膜使用的一抗为鼠抗GAPDH单克隆抗体,二抗为兔抗鼠IgG。
2 结果
2.1 慢病毒转移载体的构建和鉴定
为增加慢病毒滴度和转染率,在不影响重组蛋白表达的组织特异性和预期可以维持目的基因需要的表达水平的基础上,采用优化的基因调控因子,即选择较短的β-珠蛋白启动子,并去除β-珠蛋白IVS2和LCR中的HS1和HS4,构建了结构正确的含目的基因及报告基因的红细胞组织特异性的慢病毒载体RS9-L-ASNase-2A-mCherry-PGK-MGMT-HS4-IVS2(-)、非组织特异性慢病毒载体RRL-sEF1α-LASNase-2A-mCherry和 RRL-sEF1α-2A-mCherry(图1),并经限制性酶谱鉴定和测序分析验证。
2.2 重组慢病毒滴度检测
重组慢病毒载体分别与包装质粒psPAX2和包膜蛋白质粒pMD2.G经PEI介导瞬时共转染293T细胞获得重组慢病毒,经8500 r/min离心浓缩,用多重TaqMan PCR测得重组慢病毒RS9-L-ASNase-2A-mCherry-PGK-MGMT-HS4-IVS2(-)的滴度为(4.21±0.58)×107U/mL,RRL-sEF1α-L-ASNase-2A-mCherry的滴度为(7.6±2.5)×106U/mL,RRL-sEF1α-2A-mCherry的滴度为(2.05±0.67)×107U/mL。
2.3 重 组 慢 病 毒 RRL-sEF1α-L-ASNase-2A-mCherry在HeLa细胞中表达携带基因的检测
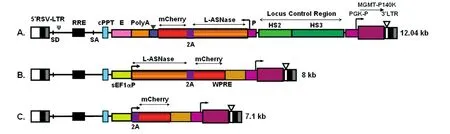
图1 慢病毒转移质粒结构示意图
为快速鉴定采用2A联体技术可实现重组慢病毒在感染细胞中同时表达目的基因及报告基因,将非组织特异性重组慢病毒RRL-sEF1α-L-ASNase-2A-mCherry按1∶10稀释后感染HeLa细胞。在激发光为587 nm、发射谱为610 nm的荧光显微镜下检测到mCherry报告基因主要在感染细胞核中表达(红色荧光)(图2B),L-ASNase基因主要在细胞质内表达,L-ASNase可与多抗结合,用键合了Alex 488的二抗可显示绿色荧光(图2C),提示mCherry与LASNase表达时自2A处自裂解,各自独立成像,无共定位(图2D)。
2.4 重组慢病毒感染MEL细胞后报告基因表达水平的测定
MEL细胞以MOI=10分别感染各包装浓缩的重组慢病毒,用流式细胞仪在指定时间点监测化疗药物BG/BCNU对重组慢病毒感染MEL细胞的选择富集效果(图3),对照为未感染慢病毒的MEL细胞。非组织特异性重组慢病毒感染MEL细胞中mCherry报告基因表达的阳性率经筛选后均有所提升,阳性细胞得到有效富集;组织特异性重组慢病毒感染MEL细胞中mCherry报告基因未表达。提示红系特异性调控因子对基因表达控制较严格。多重Taq⁃Man PCR检测结果表明,用BG/BCNU筛选16 d诱导0 d的MEL细胞中,组织特异性病毒的滴度是未经BG/BCNU筛选培养细胞的3.2倍(P=0.0025)。
红细胞特异性重组慢病毒感染的MEL细胞,经BG/BCNU富集16 d后,用5 mmol/L HMBA诱导,MEL细胞在向红系终端分化的过程中,在激发光为587 nm、发射光为610 nm的荧光显微镜下检测到mCherry报告基因在感染细胞中得到逐步增高的表达(红色荧光)(图4)。
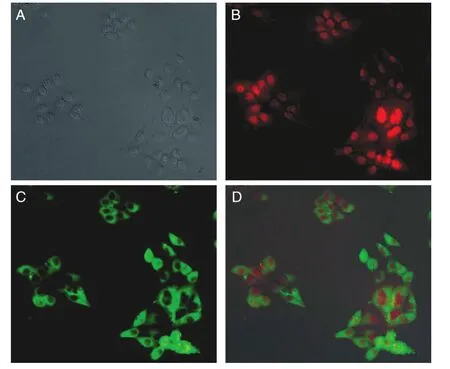
图2 重组RRL-sEF1α-L-ASNase-2A-mCherry慢病毒在HeLa细胞中的表达A:明场下感染的HeLa细胞;B:在激发光为587 nm、发射光为610 nm下感染的HeLa细胞,表达的红色单体荧光蛋白mCherry主要在细胞核内;C:L-ASNase与多抗结合,用键合了Alex 488的二抗显示绿色荧光,激发光为495 nm,发射光为519 nm,表达的L-ASNase主要在细胞质内;D:2种荧光通道与明场融合,mCherry与L-ASNase各自独立表达,无共定位
用流式细胞仪对诱导的MEL细胞进行监测,结果如图5,红细胞组织特异性重组慢病毒表达报告基因mCherry的细胞数量随诱导时间的延长而增多,非组织特异性的则相反。对照为未感染重组慢病毒,但用HMBA诱导同样时间的MEL细胞。
2.5 重组慢病毒携带目的基因及报告基因的表达鉴定
Western印迹分析显示,重组慢病毒RRL-sEF1α-2A-mCherry感染的MEL细胞裂解物中,2A肽N端的部分序列通过自裂解作用游离出来,2A肽C端的部分序列与红色单体荧光蛋白mCherry的N端结合;而重组慢病毒RS9-L-ASNase-2A-mCher⁃ry-PGK-MGMT-HS4-IVS2(-)感染的MEL细胞经BG/BCNU选择和HMBA诱导7 d后,细胞内表达的L-ASNase的C端结合了2A肽自裂解作用产生的N端部分序列(图6)。
3 讨论
在抗癌研究过程中,人们发现多种代谢异常的恶性肿瘤依赖某些外源性氨基酸,例如儿童ALL、卵巢癌、成人非霍奇金淋巴瘤不能合成L-Asn,需要从血液中吸收Asn供其生长,因而耗竭肿瘤必需氨基酸而对正常细胞不良反应最小的癌细胞选择性疗法诞生了[12]。利用L-ASNase靶向性酶解L-Asn在治疗儿童ALL中取得了显著成功,患儿5年生存率已提高到80%以上,也能明显减小非霍奇金淋巴瘤[13]。然而,由于人体内不存在天然L-ASNase,而异源LASNase在临床应用中受限的主要原因是其免疫原性,以及由此引发的过敏反应、过敏性休克及酶的免疫抑制与清除[14]。因此,降低异源酶的免疫原性,突破临床应用的局限性,成为研究人员需要解决的主要问题。
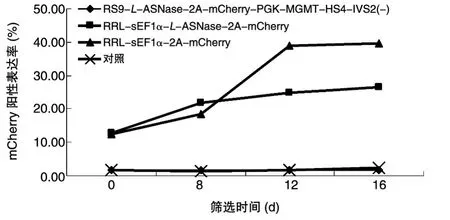
图3 流式细胞仪监测化疗药物BG/BCNU选择对MEL细胞中mCherry报告基因表达的影响
目前供临床使用的细菌源天门冬酰胺酶制剂有3种类型,一种是大肠杆菌-天门冬酰胺酶(E.coli-asparaginase,EcA)[15],第二种是聚乙二醇-天门冬酰胺 酶(polyethylene glycol-asparaginase,PEGA)[16]。EcA或PEGA是治疗ALL的一线方案。第3种欧文菌-天门冬酰胺酶(Erwinia-asparaginase,ErA)与EcA具有不同的免疫学特性,在欧洲和美国用作过敏患者的二线或三线治疗方案[17]。

图4 重组RS9-L-ASNase-2A-mCherry-PGK-MGMT-HS4-IVS2(-)慢病毒在MEL细胞中mCherry的表达情况
由于RBC的寿命和大小明显超过其他药物递送载体[18],近年来已有50多种抗感染药物、抗炎症药物、抗肿瘤药物、解毒作用的酶甚至遗传物质,或采取药物诱导的细胞内吞作用、电穿孔、蛋白质转导结构域(protein transduction domain,PTD)和低渗透压的方法把药物在体外封装到RBC内[18],或通过生物素-亲和素结合的方法把药物与RBC表面偶联的方式进入了临床研究[19]。近年来,采用基因修饰的方法使携带目的基因的造血干细胞(hematopoietic stem cell,HSC)在向终端分化成为红细胞的过程中表达而发挥治疗作用的方案,已在B型血友病研究中展开[20]。我们拟借鉴此系统,利用基因修饰后表达L-ASNase的红细胞开展恶性血液病的治疗研究。
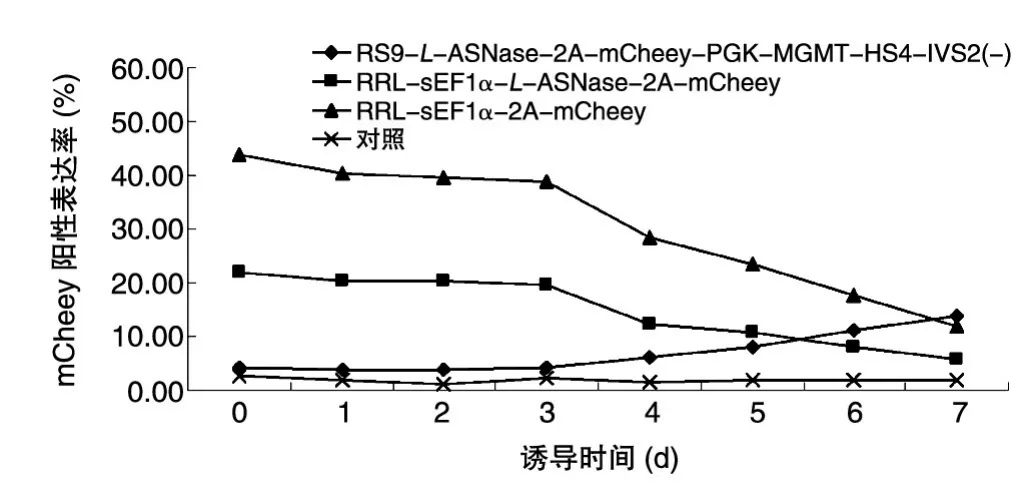
图5 流式细胞仪监测MEL细胞诱导分化期间报告基因mCherry的表达
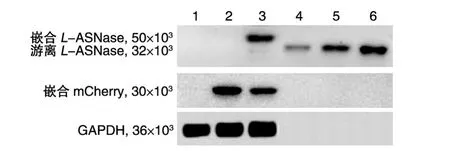
图6 重组慢病毒感染MEL细胞表达产物的免疫印迹分析
荧光显微观察结果提示,通过2A联体技术连接的L-ASNase和mCherry在细胞的不同区域表达,即mCherry不会干扰L-ASNase行使功能。流式细胞术和多重TaqMan PCR法结果显示,用50 μmol/L BG-50 μmol/L BCNU可以有效富集重组慢病毒感染的阳性细胞。由于血红蛋白是由α-珠蛋白、β-珠蛋白和血红素组成的结合蛋白,红细胞组织特异性慢病毒载体使用的是β-珠蛋白基因调控序列,可以预计当感染该重组慢病毒的MEL细胞经诱导向红系终端分化的过程中,随着血红蛋白表达的增高,红细胞组织特异性重组慢病毒携带的目的基因及报告基因的表达也将增高。非组织特异性慢病毒载体使用的是非组织特异性的sEF1α启动子,可以预计当感染该重组慢病毒的MEL细胞向红系终端分化过程中,目的基因的表达会随着血红蛋白表达的增加而逐渐受到抑制。结果显示,分别用红细胞组织特异性重组慢病毒RS9-L-ASNase-2A-mCherry-PGK-MGMT-HS4-IVS2(-)、非组织特异性重组慢病毒 RRL-sEF1α-L-ASNase-2A-mCherry和 RRL-sEF1α-2A-mCherry感染MEL细胞,经化疗药物BG/BCNU选择和HMBA诱导红细胞分化后,红细胞组织特异性重组慢病毒表达报告基因mCherry的阳性MEL细胞百分率随诱导时间的延长而增多,非组织特异性的则相反,与预计吻合。提示本研究建立的红细胞组织特异性重组慢病毒更适合于基因修饰红细胞开展恶性血液病的治疗。Western印迹结果显示L-ASNase和mCherry表达时在2A处发生了自裂解,所得表达产物与预计相符;与梯度稀释的标准品L-ASNase的灰度值相比较,在60 μg/孔总蛋白上样量中,计算出 L-ASNase量为 0.0179 U,即约为 0.3 U/mg总蛋白。参考非基因修饰方法将L-ASNase封装进RBC,给荷L5178Y淋巴瘤动物模型一次输注8 U[5],本项目预计需要制备大量携带L-ASNase的基因修饰红细胞才可能达到体内治疗效应。尽管MEL细胞模型比较经济,然而MEL细胞可使小鼠发生类似于红白血病的病征,输注由其衍生的红细胞风险较大,因此仍须选择造血干细胞进行基因修饰,我们还将继续探索。
[1] van den Berg H.Asparaginase revisited[J].Leuk Lymphoma,2011,52(2):168-178.
[2] Tarnowski G S,Mountain I M,Stock C C.Combination thera⁃py of animal tumors with L-asparaginase and antagonists of glutamine or glutamic acid[J].Cancer Res,1970,30(4):1118-1122.
[3] Jain R,Zaidi K U,Verma Y,et al.L-asparaginase:a promis⁃ing enzyme for treatment of acute lymphoblastic leukiemia[J].People J Sci Res,2012,5(1):29-35.
[4] Domenech C,Thomas X,Chabaud S,et al.L-asparaginase loaded red blood cells in refractory or relapsing acute lympho⁃blastic leukaemia in children and adults:results of the GRAS⁃PALL 2005-01 randomized trial[J].Br J Haematol,2011,153(1):58-65.
[5] Kwon Y M,Chung H S,Moon C,et al.L-Asparaginase en⁃capsulated intact erythrocytes for treatment of acute lympho⁃blastic leukemia(ALL)[J].J Control Rel,2009,139(3):182-189.
[6] Patrinos G P,de Krom M,de Boer E,et al.Multiple interac⁃tions between regulatory regions are required to stabilize an active chromatin hub[J].Genes Dev,2004,18(12):1495-1509.
[7] Kim J H,Lee S R,Li L H,et al.High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in hu⁃man cell lines,zebrafish and mice[J].PLoS One,2011,6(4):e18556.
[8] Buzina A,Lo M Y,Moffett A,et al.Beta-globin LCR and in⁃tron elements cooperate and direct spatial reorganization for gene therapy[J].PLoS Genet,2008,4(4):e1000051.
[9] 潘欣,田爱,王熙才,等.高效的红细胞特异性表达系统的建立和优化[J].转化医学杂志,2013,2(1):4-10.
[10]Pan X,Tamilselvam B,Hansen E J,et al.Modulation of iron homeostasis in macrophages by bacterial intracellular pathogens[J].BMC Microbiol,2010,10:64.
[11]Giry-Laterrière M,Verhoeyen E,Salmon P.Lentiviral vectors[J].Methods Mol Biol,2011,737:183-209.
[12]Cantor J R,Panayiotou V,Agnello G,et al.Engineering re⁃duced-immunogenicity enzymes for amino acid depletion thera⁃py in cancer[J].Methods Enzymol,2012,502:291-319.
[13]Pui C H,Carroll W L,Meshinchi S,et al.Biology,risk strati⁃fication,and therapy of pediatric acute leukemias:an update[J].J Clin Oncol,2011,29(5):551-565.
[14]Cantor J R,Yoo T H,Dixit A,et al.Therapeutic enzyme de⁃immunization by combinatorial T-cell epitope removal using neutral drift[J].Proc Natl Acad Sci,USA,2011,108(4):1272-1277.
[15]Del Casale T,Sollitti P,Chesney R H.Cytoplasmic L-aspara⁃ginase:isolation of a defective strain and mapping of ansA[J].J Bacteriol,1983,154(1):513-515.
[16]Molineux G.Pegylation:engineering improved biopharmaceuti⁃cals for oncology[J].Pharmacotherapy,2003,23(8 Pt 2):3S-8S.
[17]Pieters R,Hunger S P,Boos J,et al.L-asparaginase treat⁃ment in acute lymphoblastic leukemia:a focus on Erwinia as⁃paraginase[J].Cancer,2011,117(2):238-249.
[18]Migliaccio A R,Whitsett C,Papayannopoulou T,et al.The potential of stem cells as an in vitro source of red blood cells for transfusion[J].Cell Stem Cell,2012,10(2):115-119.
[19]Biagiotti S,Paoletti M F,Fraternale A,et al.Drug delivery by red blood cells[J].IUBMB Life,2011,63(8):621-631.
[20]Migliaccio A R,Whitsett C,Papayannopoulou T,et al.The potential of stem cells as an in vitro source of red blood cells for transfusion[J].Cell Stem Cell,2012,10(2):115-119.
猜你喜欢
家教世界(2022年34期)2023-01-08
转化医学杂志(2022年1期)2022-03-07
检验医学与临床(2021年14期)2021-07-29
国际检验医学杂志(2020年2期)2020-02-05
兽医导刊(2016年6期)2016-05-17
中国药理学通报(2015年2期)2016-01-12
热带农业科学(2015年9期)2015-10-14
中国医学科学院学报(2015年5期)2015-03-01
现代检验医学杂志(2015年1期)2015-02-06
当代畜禽养殖业(2014年12期)2014-02-27
