
两种异喹啉-7-酮的合成、表征及量化计算研究
2014-03-28 02:05顾静和王成程宇徐雨辞宋洁赵朴素
淮阴师范学院学报(自然科学版) 2014年1期
顾静和,王成,程宇,徐雨辞,宋洁,赵朴素
(淮阴师范学院化学化工学院,江苏淮安223300)
0 引言
1,8-萘二甲酰亚胺衍生物是一类发光材料,由于其具有光学材料必备的各种特性,所以被广泛应用于多种科技领域.另外,从合成的角度看,这类化合物也比较容易制备[1-5].例如,正是由于1,8-萘二甲酰亚胺衍生物能够发出很强的荧光以及具备优越的光学稳定性,使得它们在包括聚合物染色[6-7]、激光活性介质[8-9]、生物体内荧光标记物[10-11]、抗癌试剂[12]、荧光开关[13]、光发射二极管[14]、电荧光材料[15]和液晶显示器[16]等诸多方面得以应用.许多关于1,8-萘二甲酰亚胺衍生物作为荧光传感器和电-光器材的报道已经见诸报端[17-19].
关于1,8-萘二甲酰亚胺衍生物,已发表了两个化合物结构方面的信息[20-21].7H-苯并咪唑[2,1,-a]联苯[de]异喹啉-7-酮(1)和4-溴代-7H-苯并咪唑[2,1,-a]联苯[de]异喹啉-7-酮(2)是两个典型的含有1,8-萘二甲酰亚胺基团异喹啉-7-酮衍生物.虽然此前有关于它们合成方面的一些报道[5,22-24],但是,据我们所知,至今为止,还没有发现关于它们在实验和理论计算方面进一步的研究.更是没有关于化合物1和2的比较研究.事实上,不同于化合物1,在化合物2的萘环4-位上,存在一个吸电子的取代基团Br原子,这或许使得化合物2的电子结构不同于化合物1,进而影响化合物2的谱学性能.所以,当我们合成了化合物1和2,并做了详细的结构表征后,又对它们做了进一步理论研究.另外,使用实验和DFT方法,我们还对化合物1和2进行了比较研究,包括它们的电子结构和取代基团对于光谱性能的影响.希望本文呈现的研究内容对于电致发光器件的研究提供有益的信息,同时也为基于1,8-萘二甲酰亚胺衍生物的电致发光器件的研究提供一点推动作用.
1 实验与理论方法
1.1 物理测量
使用Perkin Elmer 240C元素分析仪进行碳,氢和氮的元素分析.使用Avance Mercury plus-400 instrument核磁共振仪,以TMS作内标、DMSO-d6作溶剂测试核磁共振谱.使用Nicolet 170SX红外光谱仪,在4 000~400 cm-1范围内,以KBr压片测量红外光谱.电子光谱在Shimadzu UV3100光谱仪上,以CHCCl3为溶剂测定.固体荧光光谱在F-96荧光仪上进行.
1.2 化合物1和2的制备
所有的化学试剂均为商业品,使用前未经纯化.化合物1和2的反应途径见图1.
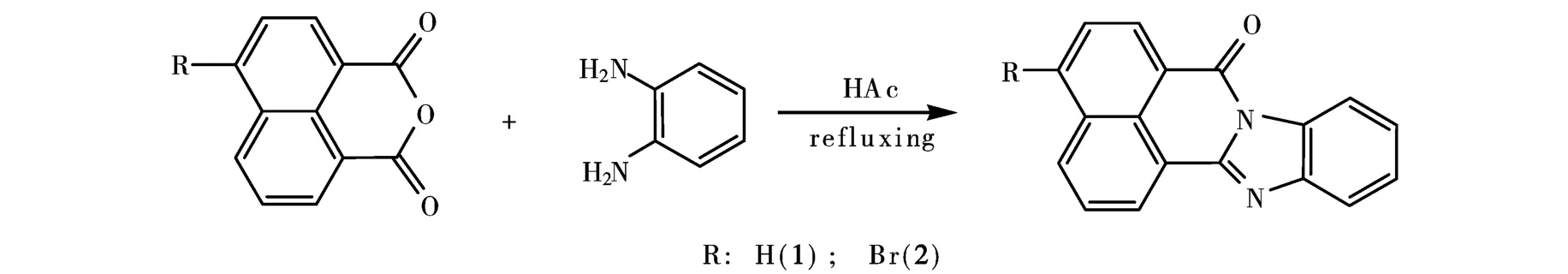
图1 化合物1和2的反应途径
7H-苯并咪唑[2,1,-a]联苯[de]异喹啉-7-酮(1)的制备:将1,8-萘酐(0.01 mmol)溶解在冰醋酸(50 mL)中,搅拌直到完全溶解,然后边搅拌边加入邻苯二胺(0.01 mmol),混合物在102~103℃进行回流.4 h之后,停止反应,用旋转蒸发器蒸去醋酸.接着,用30 mL蒸馏水稀释混合物,并加入3.0 mL盐酸(30%).将以上混合物于60℃保温1h后冷却至50℃,抽滤,洗涤得化合物1.最后,在室温下将化合物1晾干.产率90.0%.熔点205~206℃,这与文献[5]中的结果相同.元素分析结果,理论值(C18H10N2O):C,79.90;H,3.69;N,10.31%.实测值:C,79.98;H,3.73;N,10.37%.IR:ν(溴化钾压片)/cm-1:3057(m),1701(m),1653(s),1590(m),1518(m),1439(m),1341(vs),1261(s),1094(vs),1023(vs),802(s),773(m),470(m).1H NMR(DMSO,δ/ppm):7.48(d,2H,-ArH),7.85~7.92(m,2H-ArH+1H-萘环),8.32~8.70(m,5H-萘环).
4-溴代-7H-苯并咪唑[2,1,-a]联苯[de]异喹啉-7-酮(2)的制备:用和化合物1同样的方法制得化合物2.只是用4-溴-1,8-萘二钾酸酐(0.01 mmol)代替1,8-萘酐(0.01 mmol).产率95.0%.熔点224~225℃,这与在文献中[5]的结果很相似.元素分析结果,理论值(C18H9BrN2O):C,61.85;H,2.51;N,8.07%.实测值:C,61.91;H,2.59;N,8.02%.IR ν(溴化钾压片)/cm-1:3180(m),1772(w),699(vs),1569(m),1545(m),1447(m),1356(s),1228(s),1096(m),1035(m),812(m),768(m),750(m),472(m).1H NMR(DMSO,δ,ppm):7.51(d,2H,-ArH),8.03~8.11(m,2H-ArH+1H-萘环),8.41~8.82(m,4H-萘环).
1.3 计算方法
最初的分子几何模型使用MM+分子模拟和半经验AM1方法[25](HYPERCHEM 6.0,Hypercube,Ont.,Canada)获得.然后,用密度泛函方法Bemy方法[26],在B3LYP杂化函数6-311G**基组下,采用Gaussian 03软件程序[27]进行结构优化.通过振动频率的计算确定结构是稳定的(没有虚频).在最优化构型的基础上,使用含时密度泛函(TD-DFT)[28-30]方法,分别预测了化合物1和2的电子光谱.基于优化构型,实施了自然键轨道(NBO)[31]分析计算.
所有的计算都使用默认的收敛标准,在戴尔PE2850服务器和奔腾IV计算机上完成.
2 结果与讨论
2.1 优化几何构型
根据反应途径(图1),对于化合物2,可能存在如图2所示的两种构型.

图2 化合物2的两种可能的几何构型
为了选择化合物2的一种合适的几何构型用于与化合物1做对比,对化合物2a和2b的总能量在B3LYP/6-311G**水平上进行了计算,计算结果列于表1中.从表1中可以看出:化合物2a的总能量比化合物2b低0.093 kJ/mol,这说明化合物2a比化合物2b更加稳定.所以,在接下来所有讨论中,化合物2a的几何结构被选来表示化合物2的几何结构.

表1 化合物2a和2b在B3LYP/6-311G**水平上计算的总能量
用B3LYP/6-311G**方法获得了化合物1和2的优化几何构型,相应结构显示在图3中.为了让化合物1和2的比较更加直接,化合物2中除了溴原子Br(22)以外的原子编号与1保持一致.由于至今没有化合物1和2晶体结构方面的相关报道,所以我们将化合物1和2预测的几何参数与2008年[21]已经发表的化合物1的一水合盐酸盐(1·HCl·H2O)晶体结构进行了比较.化合物1和2所有的计算几何参数以及1·HCl·H2O的实验数据列于表2.
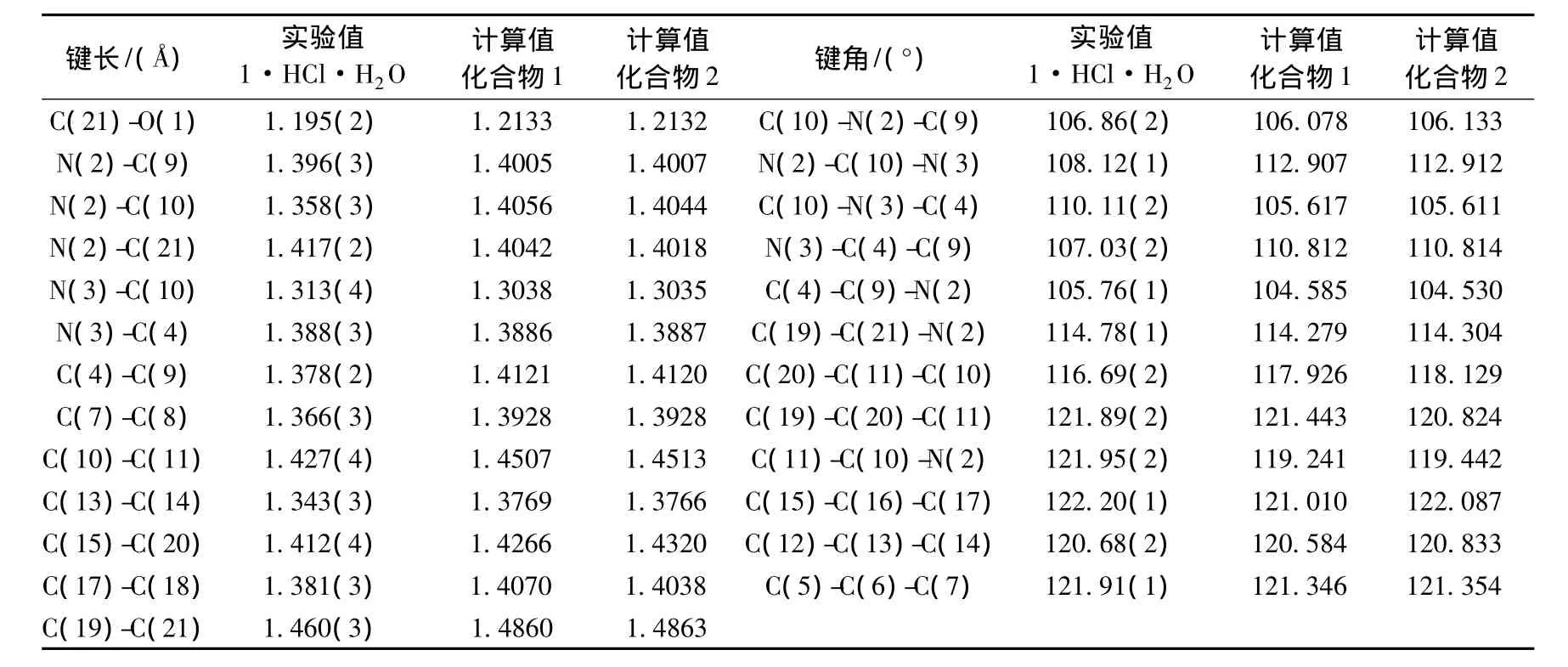
表2 1·HCl.·H2O实验键长和键角及化合物1和2在B3LYP/6-311G**水平上的理论计算值

图3 标有原子序号的化合物1和2分子结构图
从表2可以看出,对于化合物1和2,大部分预测的几何参数值都高于相应的实验值.这可能源于实验数据所描述的化合物是在固体状态下,而计算数据所对应的是气态分子.比较两个化合物预测值与实验值可以发现,它们键长和键角最大的不同主要存在于N(2)-C(10)-N(3)-C(4)-C(9)咪唑基环部分,因为在1·HCl·H2O实验值中,N(3)原子发生了质子化,并与氯原子之间存在氢键作用[21],而在计算中,化合物1和2都各自是独立的分子.对化合物1和2来说,它们与实验值最大的键长差别都发生N(2)-C(10)键,对于化合物1和2,差值分别为0.0476˚A和0.0464˚A.考虑到键角,化合物1和2与实验值最大的差别都在于C(10)-N(3)-C(4)键角,差值分别为4.499°和4.493°.比较化合物1与2二者的计算值,可以发现它们之间的差异很小,键长差别在于C(15)-C(20),差值为0.054˚A,键角差别在于C(15)-C(16)-C(17),差值为1.077°.上述的比较说明:尽管化合物2的萘环碳4位上,存在一个给电子基团溴原子,但化合物1和2的几何构型与1·HCl·H2O的晶体结构极为相似,而且B3LYP/6-311G**计算水平能为本系统研究提供令人满意的精度计算.与化合物1相比,取代溴原子并不影响化合物2主要的几何结构.
2.2 原子电荷分布
基于由B3LYP/6-311G**方法得到的化合物1和2两个优化几何构型,计算了它们的原子电荷自然布局分析(NPA).一些选定的非氢原子电荷和苯环及萘环上总的原子电荷分布分别列在表3和示于图4中.表3中还列出了化合物1和2的偶极矩值.

表3 NPA原子电荷分布及用B3LYP/6-311G**方法得到的化合物1和2的偶极矩值

图4 化合物1和2中一些非氢原子、苯环及萘环上NPA原子电荷分布(e)
从表3和图4可以看出,在化合物2的萘环碳4位引入吸电子基团Br导致了化合物2的原子电荷分布不同与1.例如,化合物2中Br原子的正电荷少于1中的H(1),化合物2中C(21)原子和C(10)原子所含有的正电荷也比1中的少.至于O(1)原子和N(3)原子,它们的负电荷在化合物2中比在化合物1中的少,而2中N(2)原子负电荷比1中的多.通过比较化合物1和2中的原子电荷值,我们可以发现,作为一个电子受体基团,Br原子把分子中其他部位的电子拉向自己,这使得化合物2的苯环所带正电荷(0.32161 e)比化合物1的苯环所带正电荷(0.31400 e)要多,化合物2中除溴以外的萘环所带负电荷(-0.03811 e)比1中除了H(1)以外的萘环所带负电荷(-0.1574 e)要少.很显然,这种原子电荷的重新分布,将导致正-负电荷中心的移动,从而使得化合物1和2的偶极矩发生变化.化合物2的偶极矩变得比化合物1的偶极矩值要小(见表3).或许,这样的原子电荷分布也将影响化合物1和2的电子光谱和荧光光谱,正如以下讨论的一样.
2.3 电子光谱
对于化合物1和2,它们的电子吸收光谱于室温下,使用在三氯甲烷(CHCl3)溶剂加以测量,实验结果列于在表4中.基于B3LYP/6-311G**方法所得优化几何构型,使用TD-DFT方法,预测了化合物1和2的电子光谱,计算结果也列于在表4中.从表4中看出,在CHCl3溶液中,化合物1和2均有3个电子跃迁峰.与化合物1中相应的峰值相比,化合物2中位于263 nm的峰值发生蓝移,另两个位于296和394 nm的峰值发生红移.在实验中,化合物2的3个峰强均弱于化合物1中相应峰强.理论计算中,使用B3LYP/6-311G**方法,化合物1和2均得到3个跃迁峰,而且与实验数据相当.化合物2中3个预测的跃迁峰强度也比化合物1中3个相应的峰强度弱.然而,与实验结果相反,与化合物1中预测的相应峰值相比,化合物2中的一个位于221 nm预测峰发生红移,而另两个位于274和339 nm的峰值发生蓝移.

表4 化合物1和2的实验和理论电子吸收光谱值
值得注意的是,对研究系统而言,化合物2中吸电子基团Br原子对电子光谱有影响.也许,就像上述研究结果所示一样,与化合物1相比,化合物2中Br原子基团的存在改变了原子电荷的分布,不仅使得电子跃迁变得更加困难,而且改变了跃迁峰的位置,降低了峰的跃迁强度.另外,将预测的理论值与实验值相比较,尽管它们之间存在一些不同点,但是所有的差别都不大,这说明B3LYP/6-311G**方法总体来说能够用来预测所研究系统的电子光谱.基于B3LYP/6-311G**优化几何的自然布局分析显示,化合物1和2的前线分子轨道主要是由p原子轨道构成.所以,与上述电子光谱相应的电子跃迁可以归属为n→π*跃迁和π→π*跃迁.图5为化合物1和2的HOMO-1,HOMO,LUMO和LUMO+1的轨道等密度表面图.由图5中可见,化合物1和2的分子轨道形状几乎相同,说明它们的电子跃迁模式是相似的.也就是说,虽然化合物2中吸电子基团Br的存在使得原子电荷发生了重新分布,并使得化合物2中的电子跃迁变得困难,但是,Br原子的存在最终并没有改变电子跃迁的模式.

图5 化合物1和化合物2中HOMO-1,HOMO,LUMO和LUMO+1轨道等密度表面图
2.4 荧光光谱
使用F96-fluorospectro荧光光谱仪测量了化合物1和2的固态荧光光谱,结果示于图6中.化合物1的发射峰在504 nm处,这属于蓝绿色范畴,半峰宽约为71 nm.化合物2的发射峰在510 nm,这也属于蓝绿色区域,半宽度大约为67 nm,与1相比,这个峰发生了一些红移.化合物2的发射峰强度比化合物1要弱.很明显,与化合物1相比,化合物2中萘环碳4位上吸电子基团Br原子的存在减弱了荧光的发射强度,并导致发射峰发生红移.也许,这种现象也是源于化合物2中原子电荷的重新分布所致.

图6 化合物1和2的固态荧光发射光谱
2.5 热力学性质
在振动分析和统计热力学基础上,化合物1和2的标准热力学函数:热容量(),熵()和焓(H0m),被计算得到,结果列于表5中.频率的校正因子为0.96,这是对于B3LYP/6-311G**方法来说的典型校正因子.

表5 B3LYP/6-311G**水平时不同温度下的热力学性质
化合物1和2的热力学性质与温度T之间的相关方程式表示如下:
对于化合物1:
对于化合物2:
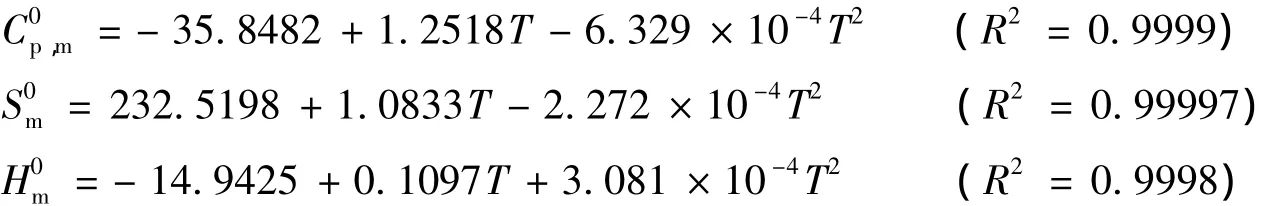
这些方程式将在进一步研究化合物1和2这两种化合物时发挥作用.
3 结论
两个异喹啉-7-酮化合物1和2已经被合成得到,并用元素分析、红外光谱、紫外光谱和荧光光谱进行了表征.DFT计算结果显示B3LYP/6-311G**计算水平在研究化合物1和2的优化几何时能够提供令人满意的精度.NPA原子电荷分布分析表明,化合物2的萘环碳4位所引入的吸电子基团Br原子在将分子中其他部分电子拉向自己时起到了重要作用,这个作用使得2的原子电荷发生了重新分布,并导致2的偶极矩发生变化.预测的化合物1和2的电子光谱与实验结果能很好地吻合.从化合物1和2的电子光谱和荧光光谱看,在化合物2的萘环碳4位引入吸电子的Br原子基团将最终改变峰的强度和峰的位置,这个信息将为人们进一步地研究基于1,8-萘二甲酰亚胺基团的电荧光材料提供有益的帮助.
[1] Tao Z F,Qian X Naphthalimide hydroperoxides as photonucleases:substituent effects and structural basis[J].Dyes Pigm,1999,43:139-145.
[2] Gan J,Chen K,Chang C P,et al.Luminescent properties and photo-induced electron transfer of naphthalimides with piperazine substituent[J].Dyes Pigm,2003,57:21-28.
[3]Refat M S,Killa H M A,Fetooh H.Spectroscopic and thermal characterization of Cu(II),Co(II),Ni(II)and Mn(II)complexes of fluorescent dye 4-N,N-dimethyl-ethanolamine-N-allyl-1,8-naphthalimide(4DMEAN)[J].J Mol Struc,2010,983:122-132.
[4] Liu F R,Du W Q,Q.Liang,et al.Synthesis of 4-aziridino[C60]fullerene-1,8-naphthalimide(C60-NI dyads)and their photophysical properties[J].Tetrahedron,2010,66:5467-5471.
[5] Tehran A.Selective preparation of fluorescent 1,8-naphthalimides using acidic alumina under microwave irradiation[J].J Chem Research(S),2001,5:485-487.
[6] Konstantinova T N,Miladinova P M.Synthesis and properties of some fluorescent 1,8-naphthalimide derivatives and their copolymers with methyl methacrylate[J].J Appl Polym Sci,2009,111:1991-1998.
[7] F Patrick L G,Whiting A.Synthesis of some polymerisable fluorescent dyes[J].Dyes Pigm,2002,55:123-132.
[8] Martin E,Weigand R,Pardo A.Solvent dependence of the inhibition of intramolecular charge-transfer in N-substituted 1,8-naphthalimide derivatives as dye lasers[J].J Lumin,1996,68:157-164.
[9] Gruzinskii V V,Kukhto A V,Shakkakh G K.Spectra of lasing efficiency in lasers with solutions of complex organic compounds[J].J Appl Spectrosc,1998,65:463-465.
[10] Jin W,Jiang J,Wang X,et al.Continuous intra-arterial blood pH monitoring in rabbits with acid base disorders[J].Respir Physiol Neurobiol,2011,177:183-188.
[11] Xie J,Chen Y,Yang W,et al.Water soluble 1,8-naphthalimide fluorescent pH probes and their application to bioimagings[J].J.Photochem Photobiol A:Chem,2011,223:111-118.
[12] Ott I,Xu Y,Qian X.Fluorescence properties and antiproliferative effects of mono-,bis-,and tris-thiophenylnaphthalimides:Results of a comparative pilot study[J].J Photochem Photobiol B:Bio,2011,105:75-80.
[13] Dai H,Xu H.A water-soluble 1,8-naphthalimide-based‘turn on’fluorescent chemosensor for selective and sensitive recognition of mercury ion in water[J].Bioorg Med Chem Lett,2011,21:5141-5154.
[14] Gan J A,Song Q L,Hou X Y,et al.1,8-Naphthalimides for non-doping OLEDs:the tunable emission color from blue,green to red[J].J Photochem Photobiol A:Chem,2004,162:399-406.
[15] Wang Y,Zhang X,Han B,et al.The synthesis and photoluminescence characteristics of novel blue light-emitting naphthalimide derivatives[J].Dyes Pigm,2010,86:190-196.
[16] Grabchev I,Moneva I,Bojinov V,et al.Synthesis and properties of fluorescent 1,8-naphthalimide dyes for application in liquid crystal displays[J].J Mater Chem,2000,10:1291-1296.
[17] Demets G J F,Triboni E R,Alvarez E B,et al.Solvent influence on the photophysical properties of 4-methoxy-N-methyl-1,8-naphthalimide[J].Spectrochim Acta A,2006,63:220-226.
[18] Grabchev I,Staneva D,Bojinov V,et al.Spectral investigation of coordination of cuprum cations and protons at PAMAM dendrimer peripherally modified with 1,8-naphthalimide units[J].Spectrochim Acta A,2008,70:532-536.
[19] Grabchev I,Dumas S,Chovelon J M.Studying the photophysical properties of a polymerizable 1,8-naphthalimide dye and its copolymer with styrene as potential fluorescent sensors for metal cations[J].Polym Adv Technol,2008,19:316-321.
[20] Jian F F,Du L,Yi W.N-(2-Hydroxybenzylideneamino)-1,8-naphthalimide[J].Acta Crystallogr,2007,E63:4748.
[21] Jian F F,Du L,Wang J.6-Oxobenz[de]isoquinolino[2,1-a]benzimidazolium chloride monohydrate[J].Acta Crystallogr,2008,E64:5321.
[22] Jiang W,Tang J N,Qi Q,et al.An experimental and computational study of intramolecular charge transfer:Diarylamino derivatives of 7H-benzimidazo(2,1-a)benz(d,e)isoquinolin-7-ones[J].Dyes Pigm,2009,80:279-286.
[23] Nakaya K,Tanaka T,Shirataki Y,et al.4-(2-Aminoethylamino)-7H-benz[de]benzimidazo[2,1-a]isoquinoline-7-one as a Highly Sensitive Fluorescent Labeling Reagent for Carnitine[J].Bull Chem SocJpn,2001,74:173-177.
[24] Lee J F,Hsu S L C.Green polymer-light-emitting-diodes based on polyfluorenes containing N-aryl-1,8-naphthalimide and 1,8-naphthoilene-arylimidazole derivatives as color tuner[J].Polymer,2009,50:5668-5674.
[25] Dewar M J S,Zoebisch E G,Healy E F,et al.Development and use of quantum mechanical molecular models 76 AM1:a new general purpose quantum mechanical molecular model[J].J Am Chem Soc,1985,107:3902-3909.
[26] Peng C,Ayala P Y,Schlegel H B,et al.Using redundant internal coordinates to optimize equilibrium geometries and transition states[J].J Comput Chem,1996,17:49-56.
[27] Frisch M J,Trucks G W,Schlegel H B.Gaussian 03 Software Package[R].Wallingford:Gaussian Inc CT,2004.
[28] Runge E,Gross E K U.Density-Functional Theory for Time-Dependent Systems[J].Phys Rev Lett,1984,52:997-1000.
[29]Petersilka M,Gossmann U J,Gross E K U.Excitation Energies from Time-Dependent Density-Functional Theory[J].Phys Rev Lett,1996,76:1212-1215.
[30] Bauernschmitt R,Ahlrichs R.Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory[J].Chem Phys Lett,1996,256:454-464.
[31] Jamorski C,Casida M E,Salahub D R.Dynamic polarizabilities and excitation spectra from a molecular implementation of time-dependent density-functional response theory:N2as a case study[J].J Chem Phys,1996,104:51345147.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
铜仁学院学报(2018年6期)2018-07-05
北京航空航天大学学报(2017年10期)2017-04-20
衡阳师范学院学报(2016年3期)2016-07-10
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
航天返回与遥感(2014年4期)2014-07-31
无机化学学报(2014年8期)2014-02-28
