
30个祖先信息位点的筛选及应用
2014-03-08 06:58李彩霞贾竟魏以梁万立华胡兰叶健
遗传 2014年8期
李彩霞,贾竟,魏以梁,万立华,胡兰,叶健
1. 公安部物证鉴定中心,北京 100038;
2. 重庆医科大学,重庆 400016;
3. 天津医科大学基础医学研究中心,天津 300070
祖先信息位点(Ancestry informative markers,AIMs)是指在不同人群之间基因频率差异非常大的多态性基因位点[1]。单核苷酸多态性(Single nucleotide polymorphisms, SNPs)由于在基因组中数量多、分布广,成为近年来筛选 AIMs位点、分析人群遗传结构的重要遗传标记之一[2,3]。一组祖先信息位点可以分析某一人群的遗传成分构成,也可以推断某一个体的祖先来源[4]。这对于避免或降低遗传学分析时由于人群分层现象导致的误差具有重要意义,同时对于法医学中推断未知 DNA样本的种族来源也具有重要意义。
不同的人群之间具有非常显著的表型差异,如肤色、发色、虹膜颜色、体内生化代谢等,这些表型的差异多与基因的多态性相关[5,6]。研究这些基因上频率分布差异显著的SNPs,通过评价族群间遗传分化指数(Fixation index between subpopulation and total population,Fst)、δ值(人群两两之间的等位基因频率的差值)等参数可获得一组 AIMs,再通过STRUCTURE遗传成分分析等可以实现人群遗传结构和个体的祖先来源推断[7,8]。本研究针对东亚、欧洲和非洲人群的遗传推断,筛选了一组祖先信息位点,建立了一套有效的检测和分析方法,以期分析DNA供者的祖先来源,为案件侦查提供线索。
1 材料和方法
1.1 研究对象
通过两个途径总共收集了14个人群的852份无关个体样本:其中,5个人群的 194份无关个体样本(3个人群124名无关个体外周静脉血和干血片以及2个人群70名无关个体的DNA样本)根据知情同意原则采集;9个人群的 658份无关个体的样本信息由 HapMap数据库下载并整理得到,根据数据库信息以及文献报道对出现三联或二联家系的人群样本(ASW、CEU、MEX和YRI)进行数据过滤筛选(表1)。所有的采集样本均有明确的人群来源信息。
1.2 方法
1.2.1 DNA提取
外周静脉血 DNA根据 QIAamp® DNA blood midi试剂盒(德国 Qiagen公司)提取;干血片 DNA根据QIAamp® DNA Mini M48试剂盒(德国Qiagen公司)提取。所有提取的DNA经过NanoDrop 2000c分光光度计(美国Thermo公司)定量。
1.2.2 AIMs位点选择
用于位点筛选的东亚、欧洲、非洲人群来自HapMap数据库,其中东亚人群包含150名个体(76名CHB和74名JPT);欧洲人群包含174名个体(104名CEU和70名TSI);非洲人群包含175名个体(77名LWK和98名YRI)。从30个表型相关基因(DCST1、SLC45A2、HMGCR、COL4A3BP、EGFR、CYP3A4、TYRP1、PDLIM1、TYR、DRD2、VDR、HMGA2、DCT、SLC24A4、OCA2、HERC2、HERC2P9、APBA2、SLC24A5、SLC12A1、MYO5C、CYP1A2、FANCA、MC1R、PRDM7、CSH1、ASIP、AHCY、TNFRSF13C和NAGA)上总共选取了282个SNP位点。这些基因与肤色、瞳孔颜色、黑色素代谢、免疫反应、泛素化以及脂质合成代谢等相关[5,6]。通过δ值、Fst值等原则评估并筛选这些 SNPs位点,具体包括:(1) 去除不符合哈迪温伯格平衡的位点(P<0.001);(2) 人群特异性位点根据δ>0.5进行筛选;(3) 通过Genepop 4.2计算3个大洲人群的Fst值,根据值的大小排序,从大到小选择位点,通过Structure软件评价位点区分效果,目标是通过最少的位点获得最佳区分效果;(4) 通过Haploview 4.2 进行位点体系的连锁平衡评价,保证体系内的位点处于连锁平衡的状态(r2<0.1)。
1.2.3 AIMs位点区分效果评价
使用筛选到的 AIMs位点区分 HapMap数据库人群,验证评价位点区分效果。共 9个人群的 658份无关个体的样本信息(YRI、LWK、ASW、CEU、TSI、GIH、MEX、CHB、JPT)。使用 Structure 2.3.4进行人群的聚类分析。使用Distruct 1.1绘制人群聚类结果图。最后将聚类结果与 HapMap所提供的样本来源信息进行比较。使用Structure软件进行。
人群聚类分析时,首先取3人群模式(即参数K= 3时)对人群的成分构成进行分析。随后根据K值的增加,观察所构建体系是否能对研究人群进行细分。
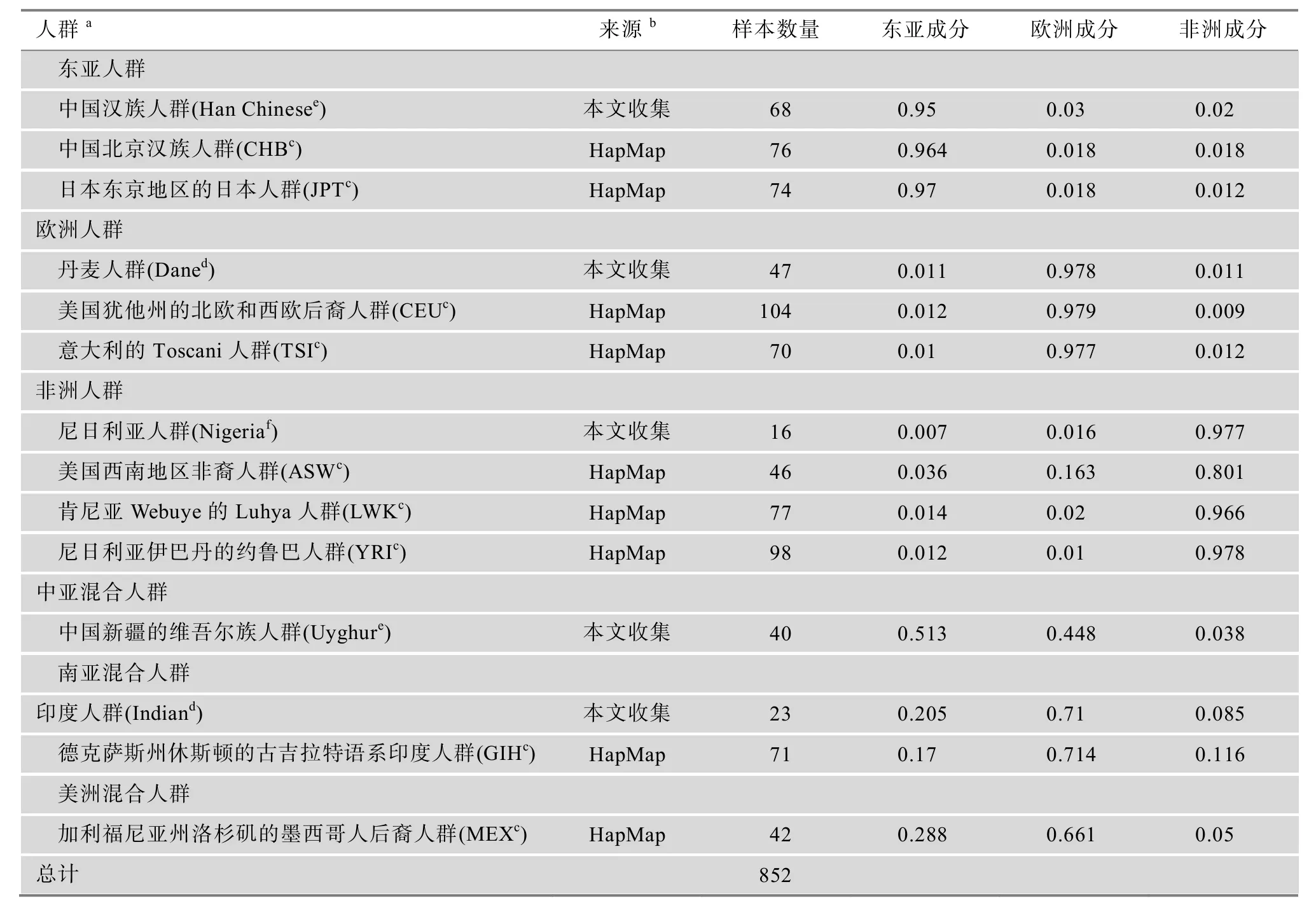
表1 人群样本及其东亚、欧、非3大人群主成分构成表
1.2.4 复合检测体系构建与样本检验
以GenomeLabTMSNPstream®基因分型系统(美国Beckman公司)构建30-plex SNPs复合检测体系。对收集的来自 5个人群的 194份样本进行分型检测(Nigeria、Dane、Indian、Uyghur、Han),随机抽取其中的50份样本进行30个位点的测序(博奥生物有限公司)验证SNP分型结果的准确性。所获得的SNP分型用STRUCTURE软件进行人群的成分构成进行分析,具体方法同1.2.3所述,分析结果同样本的已知来源进行比对,验证人群区分的准确性。
2 结果与分析
2.1 AIMs位点的筛选结果
从来自于30个基因上总共282个SNP位点中最终筛选出了 30个 AIMs位点(表 2),这组位点针对东亚、欧洲、非洲来源人群的具有显著且平衡的区分能力。并且AIMs体系中每一个SNP位点的等位基因频率在3个人群间具有较大差异。其中10个位点可用作区分东亚人群(即这些位点在非洲/东亚人群以及欧洲/东亚人群间具有较大的δ值,δ=0.652±0.096);10个位点区分欧洲人群(即这些位点在非洲/欧洲人群以及东亚/欧洲人群间具有较大的δ值,δ=0.648±0.136);10个位点区分非洲人群(即这些位点在非洲/欧洲人群以及东亚/欧洲人群间具有较大的δ值,δ=0.647±0.071)。
所有位点都符合哈迪温伯格平衡(P> 0.01),位点间连锁平衡(r2< 0.1),所有位点的Fst> 0.35。
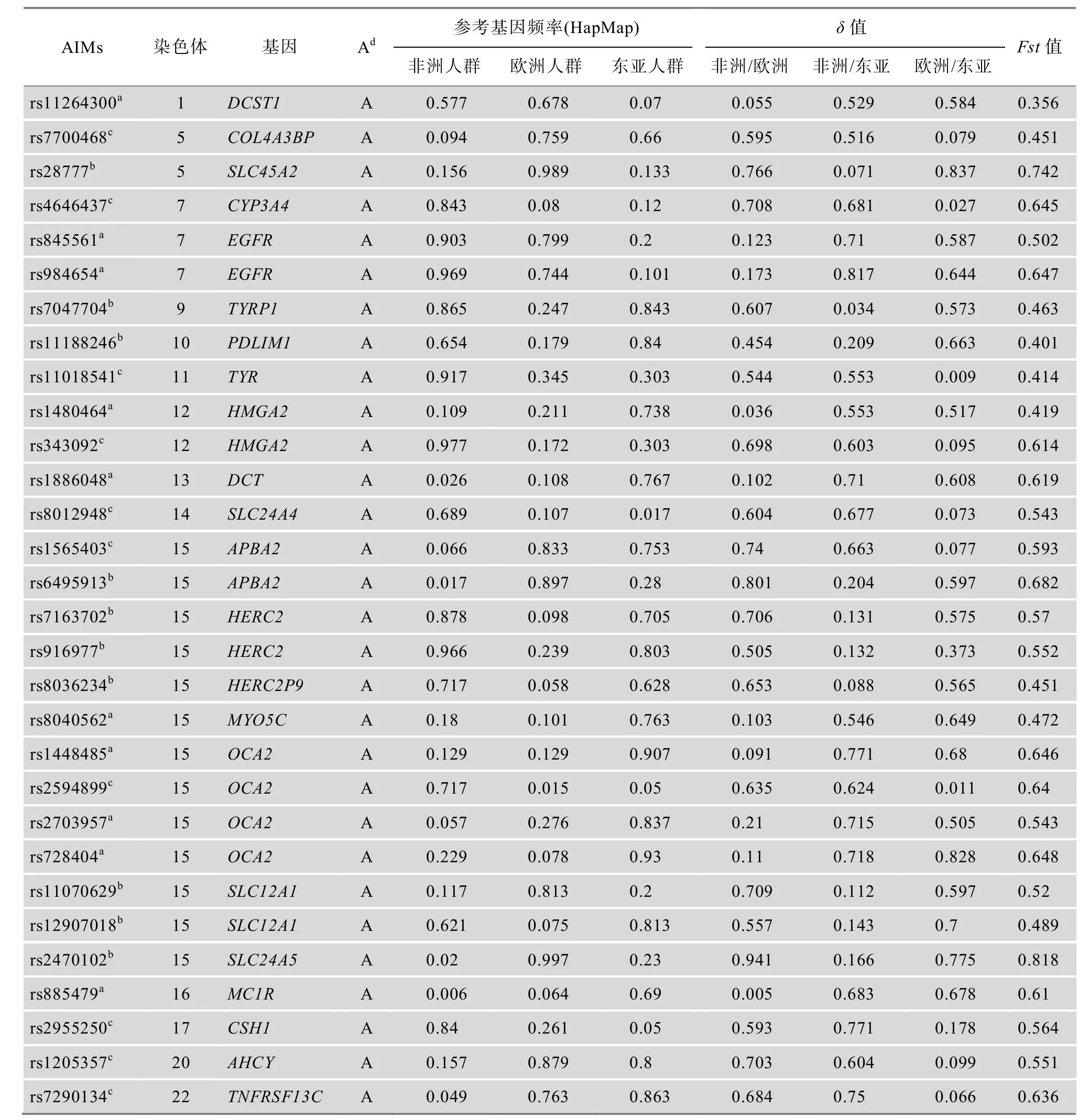
表2 30个AIMs位点的信息
2.2 AIMs位点的评估
HapMap 数据库中 9个人群的聚类结果如图 1所示。从图中可清楚的看出,9个人群在三人群模式(即参数K=3时)下,被聚类成 3个大的群体,拥有两个或3个群体成分的混合人群也可区分出来,即非洲人群(YRI和LWK)、欧洲人群(CEU和TSI)和东亚人群(CHB和 JPT)以及非洲和欧洲的混合人群(ASW)、非洲、欧洲、东亚的混合人群(GIH和MEX)。在四人群模式(即参数K=4时)下,新的人群即 GIH以独立结构显示出来,ASW、MEX人群仍以混合人群结构形式显示。若继续增加K值,未观察到新的人群以独特成分显示。
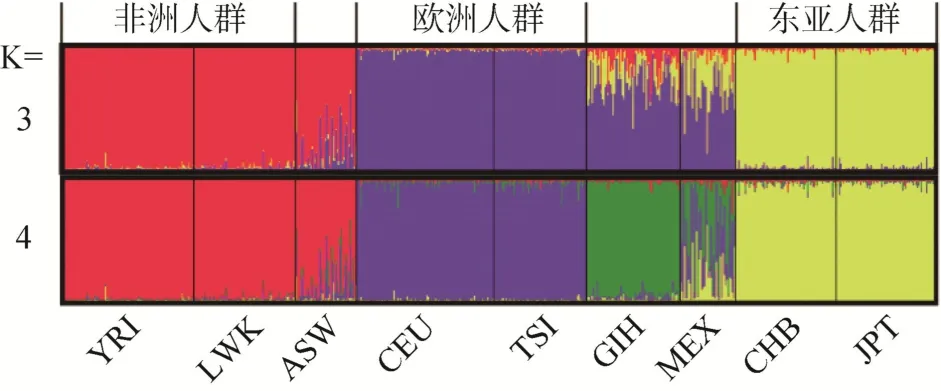
图1 HapMap数据库中9个人群聚类图
2.3 体系构建与样本检验结果
使用30-plex SNPs复合检测体系,检测5个人群(中国汉族、丹麦、尼日利亚、维吾尔族和印度)194份样本分型,共获得4762个SNP位点的分型,检出率为99%,随机抽取的50份样本测序共获得1487个SNP分型,所有测序分型结果与本研究构建体系检测结果一致。
结合HapMap的9个人群信息,将全部14个人群852份样本聚类。所有14个人群在Structure软件三人群模式(即参数K=3时)下,都可以三大人群(东亚、欧、非)成分的组合形式表示出来,即每一人群都拥有某一种或几种祖先来源成分。并且具有相同或相似祖先来源的群体,我们能观察到其结构的一致性。由表1可见,CHB、JPT、Han群体东亚成分人种主成分超过 0.95;CEU、TSI、Dane群体欧洲人种主成分超过0.97;YRI、LWK、Nigeria群体非洲人种主成分超过0.96,ASW群体中掺入欧洲人种成分;Uyghur群体为东亚人与欧洲人种成分混合;GIH、Indian、MEX以欧洲人种成分为主,少量掺入东亚人种与非洲人种成分(图2)。图3进一步展示了所有14个人群的成分构成情况。以K=3的聚类分析结果,我们将每一个人群的结构用三大人群成分的百分比构成表示。图中所标示的具体位置代表样本采集地点。从图中能清楚的看出,不同的人群拥有不同遗传主成分,相似的人群或相同来源的人群其结构具有相似性。当K=4时,与HapMap的9人群验证时的结果相似,GIH和Indian以自己的独特成分从其他人群中得以区分。若继续增加K值,未观察到新的人群以独特成分显示。通过上述分析获得的所有样本的种族结果与已知来源完全一致。
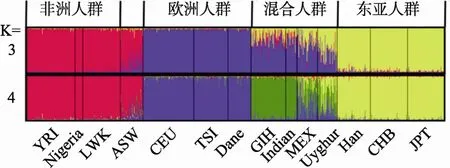
图2 14个人群的聚类图
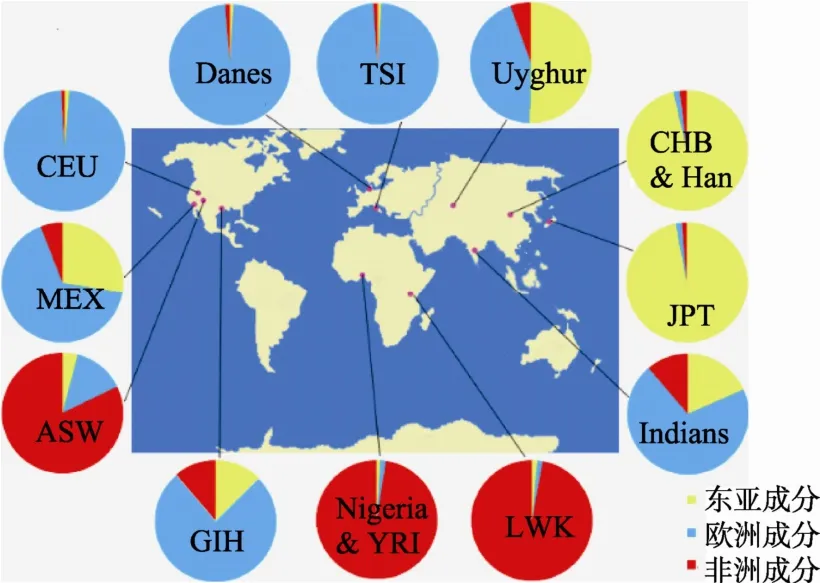
图3 14个人群的遗传成分图
3 讨 论
本研究针对东亚、欧洲、非洲三大人种,共筛选出30个AIMs位点,构建了复合SNP检测体系。该体系可以有效进行三大人群和混合人群遗传结构成分的区分。30个位点具有较高的信息含量(Fst>0.35),在所有研究人群中满足哈迪温伯格平衡及连锁平衡;并且在三个大洲来源人群的推断能力上具有平衡性(30个位点中,每10个位点为一组人群特异性位点,两两人群间的δ值>0.5)。
不同人群之间的皮肤颜色、头发颜色、虹膜颜色、代谢等特征存在较大的差异,而相关基因上的SNPs位点是导致这些差异的原因之一[5,6]。从这些基因上选择人群之间等位基因频率分布差异大的一组SNP位点可以进行人群遗传结构成分的区分[1,4,9]。本研究从 30个表型相关基因上初筛到 282个差异SNP位点。在实际检验中,位点过多会直接影响到检测体系构建的难度和体系的灵敏度,所以需进一步筛选并减少位点,同时又能保持最佳的人群区分效能。根据1.2.1中所述原则,最后筛选出30个AIMs位点。其中一些位点具有非常显著的人群特异性,例如,rs28777位于SLC45A2基因的内含子区内,等位基因A在欧洲人群中占主导,等位基因频率为0.989,而其在非洲和东亚人群中的频率分别为0.156和0.133。为了达到3个人群区分的均衡性,每个人群 AIMs位点的数量也保持一致,通过δ值将每一位点和三大人群的特征对应起来,30个AIMs位点共分为 3组。以东亚特征的 AIMs位点为例,其具有较大的非洲/东亚δ值和欧洲/东亚δ值。例如:rs885479的非洲/东亚δ值、欧洲/东亚δ值和非洲/欧洲δ值分别为0.683、0.678和0.005,说明此位点在东亚与非、欧人群具有非常大的等位基因频率分布差异,而在非、欧人群间的等位基因频率差异很小。
对应于人类学上人种分类的三分法,根据本课题的研究结果,某一人群的遗传构成可以东亚、欧、非三大人群成分所占的百分比表示;对于某一样品的祖先信息推断,可以用三大人群成分所占百分比来表示此样品的遗传成分构成从而获得其祖先来源信息。通过 HapMap人群样本和实验样本的分析可以看出,相同来源的人群呈现出遗传结构上的相似性,而混合人群具有混合的遗传成分。Han、CHB、JPT的主成分是东亚黄种人,CEU、TSI、Dane的主成分是欧洲白人,YRI、Nigeria、LWK的遗传主成分为非洲黑人。ASW是美国西南部非裔人群,虽然有一些欧洲白人成分,但其基本的构成还是以非洲黑人成分为主。而 Uyghur、MEX、Indian、GIH则为明显的混合人群,人群的遗传主成分与其地理分布具有一致性(图 3)。例如,从中国新疆收集的Uyghur人群的主成分是东亚和欧洲,同其位于欧亚大陆间的地理位置具有一致性。而位于南亚的印度人群样本则呈现出了三大人群的成分都占有一定比例的情况。但相比之下,其欧洲和非洲人群的成分较维族人群要多一些,这些都符合印度人的肤色及面部等外貌特征。另外,印度人群能以独立成分在K=4的时候显示出来,这是一个人群细分的现象,说明印度人群在遗传结构上具有其特性,这一点从K=3时的印度人群结构成分图上就能得以体现。由此可见,本研究筛选的 AIMs位点可以有效的分析三大人群及混合人群的遗传成分构成。
本研究构建的30-plex SNPs复合检测体系是基于微测序-通用芯片技术构建的,在本课题组前期研究中[10],发现该技术方法在准确性、灵敏度等方面能够满足研究需求。在本研究中,为确保分型结果准确性,我们随机抽取其中50份样本进行测序,结果表明体系检测结果与测序结果完全一致。本研究的目的主要在于筛选一组 AIMs位点,并且建立一套祖先来源推断的方法体系,所以在体系构建方面并没有做更进一步的优化。检测结果表明,所筛选的位点和建立的区分方法体系,能够很好的实现三大人群和混合人群遗传成分的分析。但是,现有检测体系的检测时间较长,需要10 h左右,而且需要专用的芯片检测装置和耗材,不利于广泛应用,在下一步研究中,我们将针对实际应用,建立更简便的适合毛细管电泳仪检测的复合体系。另外,本文筛选位点主要是针对洲际人群及其混合人群区分的,从法医学应用角度,该体系能够用于日益增多的涉外案件中个体祖先来源的推断。但是对于更多案件检验的需求,还不能完全满足遗传连锁分析中由于人群分层现象导致误差的有效控制,更进一步的亚人群的区分需要继续进行研究,例如东亚人群、中国南北方人群等。国内学者的相关报道[11~13]为下一步亚人群的区分研究奠定了良好的基础。
总之,本研究建立了30个AIMs位点的复合体系,该体系可以有效进行东亚、欧洲、非洲三大人群和混合人群的遗传主成分分析和个体的人种来源推断,相关方法可以用于法医DNA检验中未知样本的祖先来源推断,为案件提供更多侦查线索。
[1]Phillips C, Salas A, Sánchez JJ, Fondevila M, Gómez-Tato A, Alvarez-Dios J, Calaza M, de Cal MC, Ballard D, Lareu MV, Carracedo A; SNPforID Consortium. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs.Forensic Sci Int Genet,2007, 1(3–4): 273–280.
[2]Kayser M, Schneider PM. DNA-based prediction of human externally visible characteristics in forensics: motivations, scientific challenges, and ethical considerations.Forensic Sci Int Genet, 2009, 3(3): 154–161.
[3]Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW. Genetic structure of human populations.Science, 298(5602): 2381–2385.
[4]Halder I, Shriver M, Thomas M, Fernandez JR, Frudakis T.A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: utility and applications.Hum Mutat, 2008,29(5): 648–658.
[5]Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E,Cot-sapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R,Schaffner S F, Lander E S; International HapMap Consortium, Frazer KA, Ballinger DG, Cox DR, Hinds DA,Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F,Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S,Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q,Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M,Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E,Ziaugra L, Altshuler D, Shen Y,Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM,Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A,Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS,Roumy S, Sallée C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R,Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P,Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe'er I,Price A,Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF,Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L,Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A,Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR,Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G,Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS,Tsunoda T,Johnson TA, Mullikin JC, Sherry ST, Feolo M,Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD,Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K,Niikawa N, Adewole IF, Knoppers BM, Foster MW,Clayton EW, Watkin J, Gibbs RA, Belmont JW,Muzny D, Nazareth L, Sodergren E, Weinstock GM,Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM,Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT,Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M,Wallenburg JC, L’Archevêque P, Bellemare G, Saeki K,Wang H, An D, Fu H, Li Q, Wang Z, Wang R,Holden AL,Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS,Kennedy K, Jamieson R, Stewart J. Genome-wide detection and characterization of positive selection in human populations.Nature, 2007, 449(7164): 913–918.
[6]Lao O, de Gruijter JM, van Duijn K, Navarro A, Kayser M.Signatures of positive selection in genes associated with human skin pigmentation as revealed from analyses of single nucleotide polymorphisms.Ann Hum Genet,2007, 71(Pt 3): 354–369.
[7]Cockerham CC. Analyses of gene frequencies. Genetics,1973, 74(4): 679–700.
[8]Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data.Genetics,2000, 155(2): 945–959.
[9]Fondevila M, Phillips C, Santos C, Freire Aradas A, Vallone PM, Butler JM, Lareu MV, Carracedo A. Revision of the SNPforID 34-plex forensic ancestry test: Assay enhancements, standard reference sample genotypes and extended population studies.Forensic Sci Int Genet, 2013, 7(1):63–74.
[10]李彩霞, 于子辉, 贾竟, 魏以梁, 胡兰, 万立华. 法医SNP复合检测体系的构建及应用. 中国法医学杂志,2012, 27(3): 193–196.
[11]Qin P, Li Z, Jin W, Lu D, Lou H, Shen J, Jin L, Shi Y, Xu S. A panel of ancestry informative markers to estimate and correct potential effects of population stratification in Han Chinese.Eur J Hum Genet, 2014, 22(2): 248–253.
[12]Xu S, Yin X, Li S, Jin W, Lou H, Yang L, Gong X, Wang H, Shen Y, Pan X, He Y, Yang Y, Wang Y, Fu W, An Y, Wang J, Tan J, Qian J, Chen X, Zhang X, Sun Y, Zhang X, Wu B, Jin L. Genomic dissection of population substructure of Han Chinese and its implication in association studies.Am J Hum Genet, 2009, 85(6): 762–774.
[13]Zhang F, Su B, Zhang Y, Jin L. Genetic studies of human diversity in East Asia.Philos Trans R Soc Lond B Biol Sci,2007, 362(1482): 987–995.
猜你喜欢
区域治理(2022年40期)2022-11-27
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
奥秘(创新大赛)(2019年3期)2019-03-13
外国问题研究(2018年2期)2018-07-04
焊接(2017年11期)2018-01-04
中学生数理化·八年级物理人教版(2017年6期)2017-11-09
初中生世界·七年级(2017年7期)2017-09-04
百科探秘·航空航天(2016年5期)2016-11-07
