
贝特类调血脂前药与类似物的研究进展
2014-03-01 06:12:56张扬,朱昆
吉林化工学院学报 2014年3期
张 扬,朱 昆
(1.吉林化工学院化学与制药工程学院,吉林吉林132022;2.吉林大学中日联谊医院药剂科,吉林长春130033)
上世纪六十年代上市的贝特类(Fibrates,苯氧芳酸类)药物氯贝丁酯(Clofibrate)有降低甘油三酯(TG)和极低密度脂蛋白(VLDL)的作用,曾广泛应用.后经大规模、长期临床应用后发现其可导致严重的不良反应,特别是肝胆系统并发症,现已少用[1].目前,临床常用的新型贝特类药物包括吉非罗齐(Gemfibrazil)、非诺贝特(Fenofibrate)、苯扎贝特(Benzafibrate)和环丙贝特(Ciprofibrate)等,调血脂作用增强而不良反应减少,特别适用于以TG和小而密低密度脂蛋白胆固醇(sdLDL-C)升高,高密度脂蛋白胆固醇(HDL-C)降低为主要特征的动脉粥样硬化患者的血脂异常症状[2].作为应用于临床四十多年的调血脂老药,贝特类药物发挥了不可替代的作用,对其研究也从未停止过,笔者针对近年来贝特类前药及其类似物的研究进展做一综述,为今后贝特类药物的进一步开发提供依据和线索.
1 贝特类调血脂前药
1.1 前药概述
前药是一种体外无活性,或活性很小,进入体内后在酶或非酶作用下,释放出母体药物,发挥药效的物质[3].利用前药修饰可以提高药物在肠道的吸收[4-5],进而提高口服生物利用度[6-8];改善组织部分[9];提高作用靶向性[10],降低不良反应[11]等.成酯是前药常用的制备方法,贝特类药物分子中的羧基官能团为前药修饰提供了活性位点,其中氯贝丁酯与菲诺贝特就是以酯类前药的形式上市的.
1.2 非诺贝特酸前药
非诺贝特是第3代贝特类药物,口服后在酯酶作用下转化为活性代谢产物—非诺贝特酸(Fenofibric acid)而发挥疗效,见图1,其在体内的消除半衰期长达20 h,一天口服一次即可.
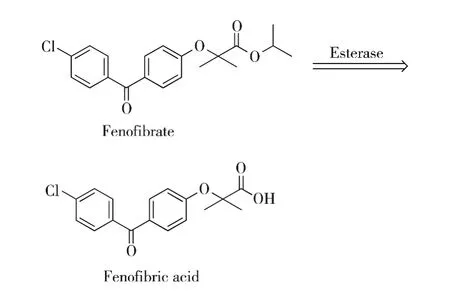
图1 非诺贝特在体内的代谢转化
由于分子中羧基的存在,非诺贝特酸在肠道的碱性环境中成离子态,限制其吸收,因此非诺贝特酸是以非诺贝特酸异丙酯(非诺贝特)的形式上市的,辅以微粉化处理,口服生物利用度提高了30%[12-13].
基于非诺贝特的设计思路,Bandgar等人[14]设计并合成了6个非诺贝特酸酯类前药1c-1h,并进行了药效学评价,50 mg/kg/d给药8天后,化合物1c和1d显示出潜在的降脂作用.在瑞士白化高血脂模型小鼠体内,化合物1c和1d可以显著地降低TG(与模型组相比,降低47%),该作用与非诺贝特相似,而降低总胆固醇(TC)的作用却优于非诺贝特.在pH=7.4的生理条件下,化合物1c(5.43)和1d(4.70)的 Log P(脂水分配系数)值与非诺贝特(4.78)接近,因此化合物1c和1d被选为候选药物进行开发.非诺贝特酸脂溶性前药1c-1h的化学结构如图2所示.
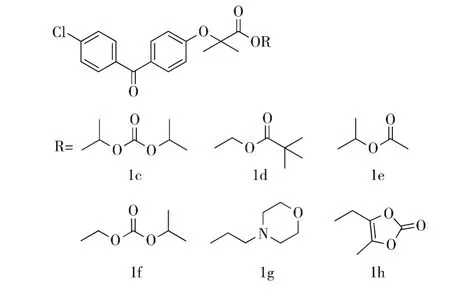
图2 非诺贝特酸脂溶性前药1c-1h的化学结构
支链低聚甘油(BGLs)是一类小分子的水溶性物质,广泛应用于药物修饰,由于其良好的水溶性和较低的分子量,在改善难溶性药物吸收方面表现出很多优于聚乙二醇(PEGs)的特性[15-16].Miyamoto等人[17]以非诺贝特酸为模型药物,BGL003(一种最简单的BGLs)为载体,通过酯键连接合成了非诺贝特酸BGL003衍生物(FFBGL),并对其进行了系统的生物学评价.试验结果表明,与非诺贝特酸相比,FF-BGL的水溶性提高了2 000倍;AUC(药-时曲线下面积)提高了3倍多;降脂活性有所提高,且未显示出明显毒性.BGL003与FF-BGL的化学结构如图3所示.

图3 BGL003与FF-BGL的化学结构
1.3 苯扎贝特前药
苯扎贝特为第二代贝特类药物,由德国BM公司开发,1979年在西德和瑞士上市,2000年在我国上市.苯扎贝特在降低TG、升高HDL-C方面效果明显,主要用于高TG血症、高胆固醇血症、混合型高脂血症的治疗[18].受非诺贝特酸酯类前药启发[14],Bandgar等人[19]又对苯扎贝特酯类前药进行了研究,合成了7个苯扎贝特衍生物1-7,并对其进行了较为系统的生物学评价.药效试验结果表明,在瑞士白化高血脂模型小鼠体内,50 mg/kg/d给药 8天后,化合物2,3,6,7在降低TG方面优于苯扎贝特,其中化合物7活性最高,其对TG的降低率高达30%;在降低总胆固醇(TC)方面,苯扎贝特和其前药均未表现出优势.体外研究表明,化合物 2,3,5,6,7 在 pH=7.4的生理条件下的Log P值均大于苯扎贝特,这与药效试验结果符合,推测可能由于成酯后提高了母体药物的脂溶性,改善了生物利用度,进而提高了活性.其中化合物7(Log P=3.1),由于其活性最高,被选为候选药物,作进一步开发.苯扎贝特与其衍生物1-7的化学结构如图4所示.
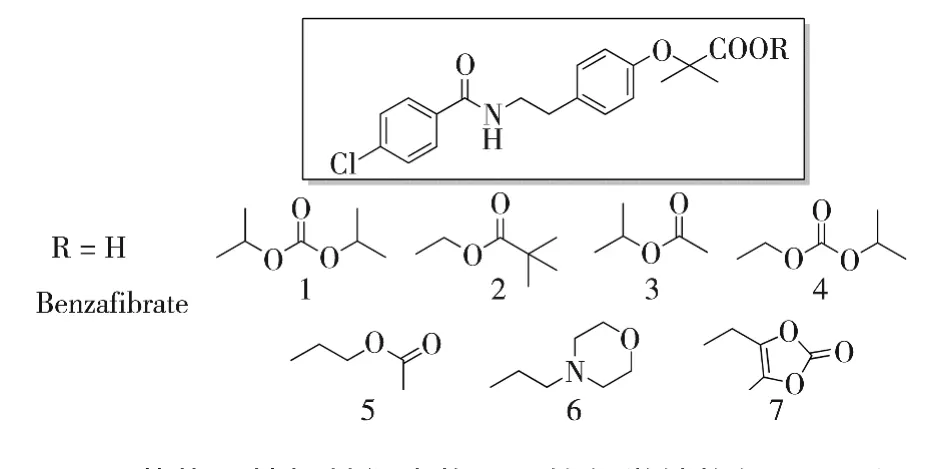
图4 苯扎贝特与其衍生物1-7的化学结构如图5所示
1.4 吉非罗齐挛药
挛药(Codrug)是指应用拼合原理(Combination principles)将两种药物的结构拼合在一个分子中,或将两者的药效基团兼容在一个分子中,所组成新的杂交分子,新形成的杂交分子或兼具两者的性质,强化药理作用;或使两者取长补短,发挥各自的药理活性,起到协同作用[20].广义而言,挛药属于前药,都是应用了药物潜伏化原理,新分子必须经过体内转化才能发挥药理作用,故挛药又称协同前药(Mutual prodrug).区别在于前药的载体无活性,而挛药的载体多是活性分子[21].
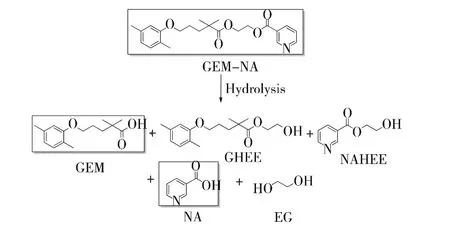
图5 GEM-NA的水解产物
吉非罗齐(Gemfibrazil,GEM),又名吉非贝齐,是1982年在美国上市的贝特类药物,能降低血浆中富含TG的脂蛋白,尤其是VLDL,升高HDL,适用于严重高脂蛋白血症[22].烟酸(Nicotinic acid,NA),又称维生素B3,是人体必需的维生素之一,其在人体内转化为烟酰胺,参与体内脂质代谢,降低血浆中 TG[23].Qandil等人[24]应用拼合原理将吉非罗齐和烟酸通过连接臂组合在一个分子中,合成了吉非罗齐-烟酸挛药(GEM-NA),并对其进行了体外水解研究.试验结果表明,在胃肠道pH条件下GEM-NA具有良好的稳定性,半衰期随着pH的增大而缩短,介于42-289天.尽管没有报道GEM-NA的体内研究结果,但是贝特类挛药的设计思路还是值得借鉴的.GEM-NA的水解产物如图5所示.
2 贝特类调血脂类似物
2.1 贝特类药物作用靶点
过氧化体增殖物激活型受体 (peroxisome proliferators activated receptors,PPARs)主要有三类亚型:PPARα、PPARγ 和 PPARδ,其中,激活PPARα可以触发系列与调控脂类物质代谢有关的生理、生化反应,进而降低血浆内TG和游离脂肪酸水平,同时升高HDL水平[25].经典贝特类药物的作用靶点就是PPARα.近年来研究表明,除PPARα外,PPARγ和PPARδ也与代谢性疾病有关.PPARγ通过调控脂代谢基因,而在脂肪酸贮存与糖代谢方面等发挥重要作用;PPARδ通过调节脂肪酸异化作用,胰岛素敏感性和组织能量平衡等发挥作用[26-27].
除了前药修饰,近年来,贝特类药物在围绕其靶点的类似物开发方面也取得了一些研究成果,既包括经典的选择性 PPARα激动剂,也包括PPARα/γ/δ混合型激动剂.在这一部分中,笔者将针对贝特类调血脂类似物的研究进展做一简单介绍.
2.2 选择性PPARα激动剂
经典的贝特类药物对PPARα选择性较高,但对其亲和力较弱,近年来,基于PPARα的激动剂开发主要围绕如何提高受体亲和性展开的,其中意大利学者Amoroso课题组做了大量的探索性研究,并以氯贝特为模型药物,总结了高活性PPARα 激动剂的构效关系[28-30],见图6.
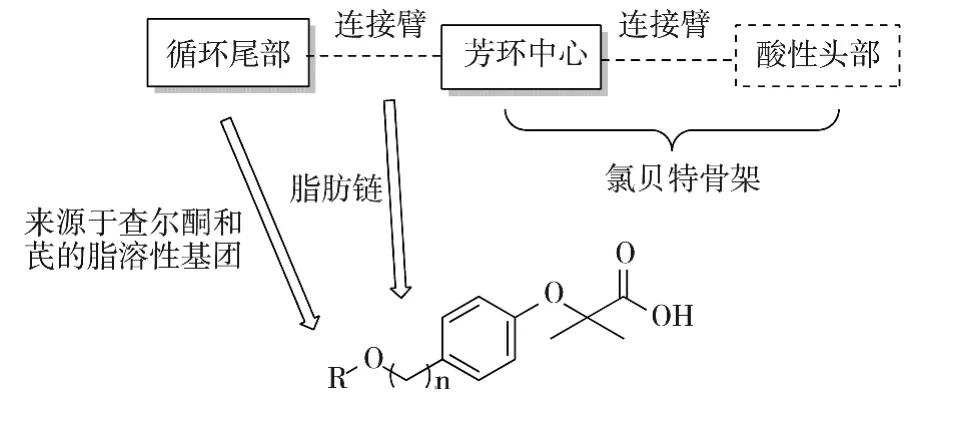
图6 高活性PPARα激动剂的构效关系
与经典贝特类药物类似,高活性PPARα激动剂由三部分组成:(1)带有脂溶性重复结构片段的循环尾部,特别是来源于查尔酮、芪等天然产物的结构片段;(2)芳环中心;(3)酸性头部;(4)n为1-3的整数;(5)三部分之间通过脂肪链连接.
在此构效关系指导下,Amoroso等人[31]继续对高活性PPARα激动剂进行了探索:保留贝特类骨架的前提下,对尾部R基团和连接臂长度进行系列变换,设计并合成了化合物1-21,见图7.
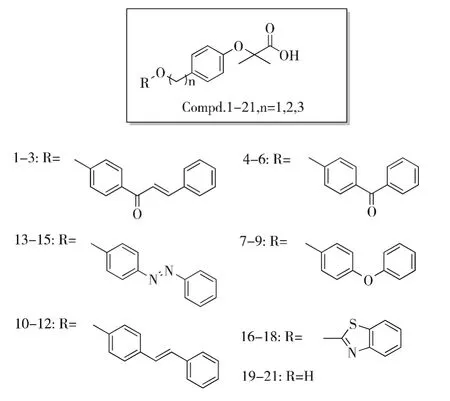
图7 化合物1-21的化学结构
活性测试表明,化合物12(PPARα,EC 50=0.8 μM;PPARγ,EC50=9.9 μM)和 14(PPARα,EC 50=0.6 μM;PPARγ,EC50=1.4 μM)不仅对PPARα活性较高,对 PPARγ也有一定活性,为PPARα/γ双重激动剂.在CPT1A基因表达测试中,化合物14可以促进CPT1A基因mRNA的表达水平,并成剂量依赖型.
2.3 PPARα/δ激动剂
研究表明,单纯激动PPARα仅能调节血脂,而同时激动PPARγ/δ理论上则可以起到协同降低血糖的作用.由于高血脂患者多数都不同程度的高血糖,反之亦然,因此,“多靶点协同治疗”概念对于代谢性疾病的治疗具有较大的现实意义.世界各国学者也在不断开发PPARα/γ/δ混合型激动剂,其中以日本学者Yuichi Hashimoto领导的课题组[32]在混合型PPARα /δ 激动剂方面做了较大贡献,开发了以3,4-二取代苯丙酸为骨架的PPARα/δ双重激动剂,并总结了这类化合物的构效关系,见图8,为3,4-二取代苯丙酸类PPARα/δ激动剂的开发提供依据.
与PPARα激动剂类似,PPARα/δ激动剂也可以划分为3部分:(1)苯丙酸基本母核;(2)带有酰胺键的连接臂;(3)具有大位阻的脂溶性取代基.区别在于,取代基赋予更多变换,集中体现在R1、R2和R3上.在此构效关系指导下,经过筛选,得到具有开发潜力的PPARα/δ激动剂TIPP-401,见图 9.活性测试结果表明,TIPP-401对PPARα的EC50=10 nM,对 PPARδ的 EC50=12 nM,二者活性非常接近,且都为纳摩尔级,具有较好的成药性.
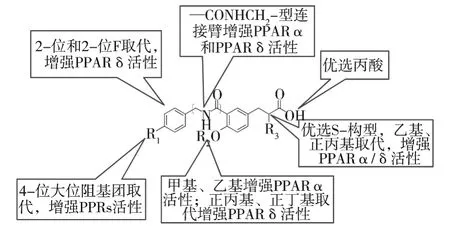
图8 3,4-二取代苯丙酸类 PPARα/δ激动剂的构效关系
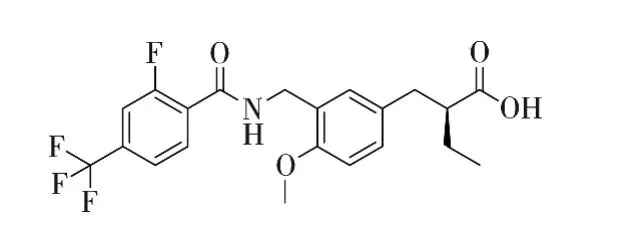
图9 化合物TIPP-401的化学结构
今年来,基于PPARs的药物研究层出不穷,筛选出了多个具有开发潜力的先导物,如TIPP-401等.遗憾的是PPARγ激动剂的研究却相对滞后,笔者将对其最新研究成果进行追踪,结果将另文报道.
3 展 望
前药修饰是一种成功率高,开发周期短的新药研究方法,大约5-7%的上市药物可以定义为前药[33].贝特类药物分子中的羧基官能团为前药修饰提供了活性位点,上述前药实例都是酯类衍生物.对于口服药物的吸收而言,分子量和Log P扮演着重要的角色,Lipinski归纳的“类药5规则”中提出分子量低于500,Log P小于5的口服药物才能被机体充分吸收[34].虽然“类药5规则”只是普遍性规律,仅限经被动扩散方式吸收的药物分子,却为前药开发提供了很好的设计指导.目前,互联网上也有很多预测Log P的免费在线软件,如 Molinspiration、ALOGPS 2.1 等.今后开发贝特类前药时,可以通过分子量和Log P进行初步的成药性判断,反过来指导设计,减少盲目性.另外,由于高血脂是导致很多心脑血管疾病的“元凶”,心脑血管疾病都伴有不同程度的高脂血症,因此应用拼合原理将贝特类药物与其他心脑血管药物缀合成挛药,发挥协同作用,也是一个颇具前景的开发方向.与前药相比,针对贝特类药物受体PPARs的类似物开发更具有合理性,特别在“多靶点,协同治疗”这一块更是筛选出多个有潜力的先导物,是对经典贝特类药物的一个活性延伸.
4 结 论
总之,在这个心脑血管疾病高发的年代,调血脂药物的开发始终是医药研究热点,而像贝特类这些经过充分临床验证的老药,更值得我们医药工作者的关注,老药的二次开发是一条符合国情、低风险的新药开发之路.
[1] 杨宝峰.药理学[M].8版.北京:人民卫生出版社,2013.
[2] Fruchart,J.C.;Sacks,F.M.;Hermans,M.P.,et al.The residual risk reduction initiative:a call to action to reduce residual vascular risk in dyslipidaemic patient[J].Diab.Vasc.Dis.Res.,2008,5(4):319-335.
[3] 张扬,肖振晶,董卫权.姜黄素前药的研究进展[J].吉林化工学院学报,2011,28(11):22-27.
[4] Kubo,S.H.;Cody,R.J.Clinical pharmacokinetics of the angiotensin converting enzyme inhibitors.A review[J].Clin.Pharmacokinet.1985,10(5):377-391.
[5] Mizuno,N.;Niwa,T.;Yotsumoto,Y.,et al.Impact of drug transporter studies on drug discovery and development[J].Pharmacol.Rev.2003,55(3):425-461.
[6] Baudy,R.B.;Butera,J.A.;Abou-Gharbia,M.A.,et al.Prodrugs of perzinfotel with improved oral bioavailability[J].J.Med.Chem.2009,52(3):771-778.
[7] Xie,Q.;Wang,X.;Jiang,Z.;Qiu,Z.Design,synthesis,and bioavailability evaluation of coumarin-based prodrug of meptazinol[J].Bioorg.Med.Chem.Lett.2005,15(22):4953-4956.
[8] Kahns,A.H.;Moss,J.;Bundgaard,H.Improved oral bioavailability of salicylamide in rabbits by a 1,3-benzoxazine-2,4-dione prodrug[J].Int.J.Pharm.1992,78(1-3),199-202.
[9] Zhang,Y.;Yang,Y.;Zhao S.,et al.Phenolic esters of O-desmethylvenlafaxine with improved oral bioavailability and brain uptake[J].Molecules.2013,18,14920-14934.
[10] Horn,A.S.;Kelly,P.;Westerink,B.H.,et al.A prodrug of ADTN:Selectivity of dopaminergic action and brain levels of ADTN[J].Eur.J.Pharmacol.1979,60(1),95-99.
[11] Bandgar,B.P.;Sarangdhar,R.J.;Viswakarma,S.,et al.Synthesis and biological evaluation of orally active prodrugs of indomethacin[J].J.Med.Chem.2011,54(5):1191-1201.
[12] Keating,G.M.;Ormrod,D.Micronized fenofibrate:an updated review of its clinical efficacy in the management of dyslipidemia[J].Drugs.2002,62(13):1909-1944.
[13] Miller,D.B.;Spence,J.D.Clinical pharmacokinetics of fibric acid derivatives(fibrates).Clin.Pharmacokinet[J].1998,34(2):155-162.
[14] Bandgar B.P.,Sarangdhar R.J.,Khan F.,et al.Synthesis and biological evaluation of orally active hypolipidemic agents[J].J.Med.Chem.2011,54(16):5915-5926.
[15] Ishihara,A.;Yamauchi,M.;Kusano,H.,et al.Preparation and properties of branched oligoglycerol modifiers for stabilization of liposomes[J].Int.J.Pharm.2010,391(1-2):237-243.
[16] Miyamoto,L.;Watanabe,M.;Kono,M.,et al.Cytotoxicity evaluation of symmetrically branched glycerol trimer in human hepatocellular carcinoma HepG2 cells[J].J.Toxicol.Sci.2012,37(5):1059-1063.
[17] Miyamoto L.;Watanabe M.;Taoka C.,et al.A novel prodrug strategy for extremely hydrophobic agents:Conjugation to symmetrically branched glycerol trimer improves pharmacological and pharmacokinetic properties of fenofibrate[J].Mol.Pharm.2013,10,2723-2729.
[18]吴洁,固旭,李东等.降脂药苯扎贝特合成工艺改进[J].中国新药杂志.2010,19(4):311-312.
[19] Bandgar B.P.;Sarangdhar R.J.;Fruthous K.,et al.Synthesis and biological evaluation of ester prodrugs of benzafibrate as orally active hypolipidemic agents[J].Eur.J.Med.Chem.2012,57:217-24.
[20]刘敏,章文军,高宁.拼合原理及其在新药设计中的应用[J].化学试剂,2009,31(10):795-797.
[21] Abdel-Azeem A.Z.;Abdel-Hafez A.A.;El-Karamany G.S.,et al.Chlorzoxazone esters of some non-steroidal anti-inflammatory(NSAI)carboxylic acids as mutual prodrugs:design,synthesis,pharmacological investigations and docking studies[J].Bioorg.Med.Chem.2009,17(10):3665-3670.
[22] Todd P.A.;Ward A.Gemfibrozil.A review of its pharmacodynamicand pharmacokinetic properties,and therapeutic use in dyslipidaemia[J].Drugs.1988,36(3):314-339.
[23] Zeb Shah T.;Ali A.B.;Ahmad Jafri S.,et al.Effect of nicotinic acid(Vitamin B3 or Niacin)on the lipid profile of diabetic and non-diabetic rats[J].Pak.J.Med.Sci.2013,29(5):1259-1264.
[24]Qandil A.M.;Rezigue M.M.;Tashtoush B.M.Synthesis,characterization and in vitro hydrolysis of a gemfibrozil-nicotinic acid codrug for improvement of lipid profile[J].Eur.J.Pharm.Sci.2011,43(3):99-108.
[25] Fruchart J C.Peroxisome proliferator-activated receptor-alpha(PPARα):at the crossroads of obesity,diabetes and cardiovascular disease[J].Atherosclerosis.2009,205(1):1-8.
[26] Lehrke M,Lazar M A.The many faces of PPARγ[J].Cell.2005,123(6):993-999.
[27] Wagner K D,Wagner N.Peroxisome proliferator-activated receptor beta/delta(PPARβ/δ)acts as regulator of metabolism linked to multiple cellular functions[J].Pharmacol.Ther.2010,125(3):423-435.
[28] Giampietro L,Ammazzalorso A,Giancristofaro A,et al.Synthesis and biological evaluation of 2-heteroarylthioalkanoic acid analogues of clofibric acid as peroxisome proliferator-activated receptor α agonists[J].J.Med.Chem.2009,52(20):6224-6232.
[29] De Filippis B,Giampietro L,Giancristofaro A,et al.Synthesis and biological evaluation of gemfibrozil chiral analogues as potential PPARα agonists[J].Lett.Drug Des.Disc.2011,8(2):154-158.
[30] De Filippis B,Giancristofaro A,Ammazzalorso A,et al.Discovery of gemfibrozil analogues that activate PPARα and enhance the expression of gene CPT1A involved in fatty acids catabolism[J].Eur.J.Med.Chem.2011,46(10):5218-5224.
[31] Giampietro L,D’Angelo A,Giancristofaro A,et al.Synthesis and structure-activity relationships of fibratebased analogues inside PPARs[J].Bioorganic Bioorg.Med.Chem.2012,22(24):7662-7666.
[32] Miyachi H,Hashimoto Y.Structural Development Studies of Subtype-Selective Ligands for Peroxisome Proliferator-Activated Receptors(PPARs)Based on the 3,4-Disubstituted Phenylpropanoic Acid Scaffold as a Versatile Template[J].PPAR Res.2008,2008:689-859.
[33] Rautio J.;Kumpulainen H.;Heimbach T.,et al.Prodrugs:design and clinical applications[J].Nat.Rev.Drug Discov.2008,7(3):255-272.
[34] Duchowicz P.R.;Talevi A.;Bellera C.,et al.Application of descriptors based on Lipinski's rules in the QSPR study of aqueous solubilities[J].Bioorg.Med.Chem.2007,15(11):3711-3719.
猜你喜欢
山西化工(2019年1期)2019-03-28 11:57:58
中成药(2018年10期)2018-10-26 03:41:22
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
大理大学学报(2018年2期)2018-04-21 07:00:38
校园英语·下旬(2018年12期)2018-02-26 12:48:32
山东医药(2017年5期)2017-04-05 05:18:49
医学研究杂志(2015年5期)2015-06-10 06:43:26
华人时刊(2014年2期)2014-03-28 00:45:01
眼科新进展(2014年3期)2014-03-08 07:20:26
中国神经精神疾病杂志(2013年1期)2013-03-11 20:23:36
