
一例婴儿型Sandhoff病家系的基因诊断与产前诊断
2013-11-29 06:45吴桐菲李溪远刘玉鹏宋金青杨艳玲
浙江大学学报(医学版) 2013年4期
吴桐菲,李溪远,王 峤,刘玉鹏,丁 圆,宋金青,张 尧,杨艳玲
(北京大学第一医院儿科,北京 100034)
GM2神经节苷脂沉积病为较罕见的溶酶体病,因己糖胺酶缺乏导致GM2神经节苷脂沉积所致,为常染色体隐性遗传病[1]。Sandhoff病由 Konrad Sandhoff于1968 年首先描述[2],是一种罕见的GM2神经节苷脂贮积症,约占GM2神经节苷脂贮积症的7%[3]。己糖胺酶酶有A(Hex A)和 B(Hex B)两种主要同功酶,Sandhoff病为己糖胺酶A和B的共同缺陷,不仅导致GM2神经节苷脂在脑中进行性沉积,同时导致糖脂、糖蛋白及低聚糖在脑与内脏中沉积,临床表现类似黑矇性痴呆,但常伴随内脏受累[4]。己糖胺酶酶A由一条α肽链和一条β肽链组成,己糖胺酶酶B由两条β肽链组成,β肽链基因突变时,则引起两种酶的同时缺乏。编码 β 肽链的基因 HEXB(EC3.2.1.52)位于5q13.3,全长 36 145 bp,含 14 个外显子[5]。
迄今国内关于 Sandhoff病的报道较少[4,6],临床诊断困难,目前尚无有效的治疗方法。据国外文献报道,加拿大萨斯喀彻温省Sandhoff病发病率较高[7],多数国家发病情况不明。随着对溶酶体病的关注,我国关于神经节脑苷脂病的散发临床病例研究增多[4],但缺乏基因研究及产前诊断的经验。本研究拟就一例Sandhoff病患儿的诊断经过、分子遗传学研究结果及该家系新生婴儿的产前诊断进行分析。
1 病例资料
1.1 先证者诊疗经过 临床表现:患儿男,1岁5个月时主因“精神运动发育倒退,抽搐”来本院就诊。患儿生后8个月内未见明显异常,3个月竖头,4个月翻身,7个月会独坐,但易惊,畏怕声音,咽部痰多,打鼾,易呛咳。8个月后发育倒退,无力,精神萎靡。1岁时颅脑CT扫描未见明显异常病变。脑MRI示两侧脑沟增宽加深,两侧额颞部蛛网膜下腔增宽。血液氨基酸及酯酰肉碱谱分析未见异常。尿液有机酸分析未见异常。染色体核型正常。当地医院曾诊断“脑发育不良,脑性瘫痪?”,功能训练无效,患儿病情进行性加重,反复上呼吸道感染,1岁时不能坐,不会翻身,不认人,不会说话。1岁4个月起惊跳后眨眼,四肢阵挛样抽搐。
出生史及家族史:患儿系第一胎第一产,足月顺产出生,出生体重3 200 g。患儿父母健康,祖籍浙江,双方无类似疾病家族史。
体格检查:头围48 cm,体重10.5 kg,身长83 cm,四肢肌张力低下,反射亢进,脊柱后凸。双侧眼底可见樱桃红斑,视神经萎缩。
X线检查:骨龄正常,双侧腕骨指骨及腰椎骨质未见异常,胸腰椎后凸。
脑MRI(图1):双侧丘脑 T1信号增高,T2WI信号减低;双侧豆状核可见多发小片状长T1、长T2信号,T2FLAIR呈稍高信号,DWI未见异常高信号;双侧大脑半球白质弥漫性T2WI信号增高,双侧额叶白质髓鞘化不良,T1WI呈低信号。
常规化验:血液及尿液常规化验未见异常,肝肾功能、肌酶、碱性磷酸酶正常。
脑电图:轻度异常,多量4~7 c/s低至中幅θ波及少至中量2~3.5 c/s低至100微伏δ波各导弥散分布,两侧对称;少量低幅快波夹入各导。
心电图及超声心动图:未见异常。
肌电图:双侧第一背侧骨间肌募集反应较差。
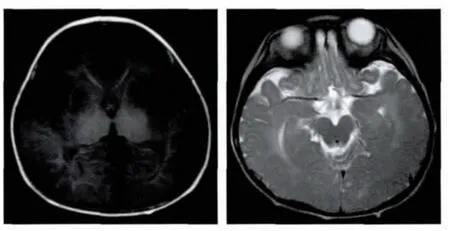
图1 Sandhoff病患儿1岁5个月时脑MRI扫描结果Fig.1 Cranial MRI of the patient with Sandhoff disease at the age of 17 months
腹部超声检查显示肝脏轻度增大,右叶最大斜径7.6 cm,肋下1.3 cm 可探及;肠胀气。
外周血白细胞溶酶体酶活性测定(荧光分光光度测定法)[8-9]:患儿己糖胺酶A+B酶活力为0(正常值为41.9 ~135.1 nmol/mg/h),严重缺陷;己糖胺酶A酶活力为1.5 nmol/mg/h(正常值2.8~9.5 nmol/mg/h),酸性 β -半乳糖苷酶活性正常(33.8 nmol/mg/h,12.6 ~52.7 nmol/mg/h)。
HEXB基因分析(图2):在知情同意的前提下抽取患儿及其父母外周血,采用Miller盐析法提取白细胞基因组DNA[10]。由NCBI网站下载 HEXB基因组全序列(http://www.ncbi.nlm.nih.gov/nuccore/NG_009770.1?from=5001&to=41145&report=genbank),GenBank ID为3074。获取HEXB基因的相关信息,并应用Primer5软件设计PCR引物(表1),采用双向测序法进行基因序列分析。

表1 HEXB基因外显子序列扩增引物Table 1 Primers for the amplification of HEXB gene
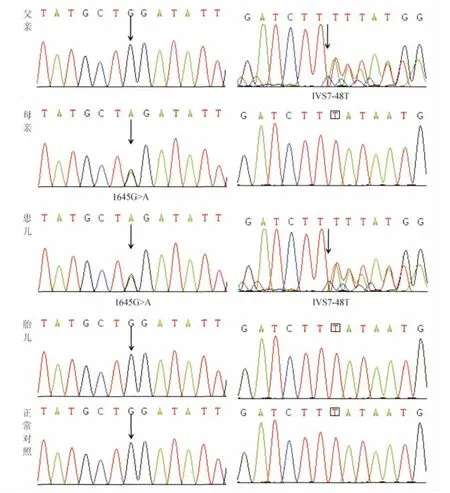
图2 Sandhoff病家系及胎儿HEXB基因测序图Fig.2 Partial sequence chromatograms of HEXB gene of the family affected by Sandhoff disease and the fetus
结果证实患儿HEXB基因外显子14存在c.1645G>A错义突变及内含子7存在c.IVS7-48T缺失(图2),符合 Sandhoff病诊断。其母亲携带c.1645G>A突变,父亲携带IVS7-48T缺失。
治疗与随访:目前患儿2岁3个月,进行性衰竭,反复呼吸道感染,消瘦,频繁抽搐,吞咽困难,双眼失明,四肢及肌张力增高,痉挛性瘫痪状态。多种抗癫痫治疗无效。
1.2 下一胎同胞的产前诊断 患儿母亲于第2次妊娠18周时来院,希望进行胎儿产前诊断,于妊娠19周时抽取羊水,分取羊水细胞,进行HEXB基因诊断,未检出c.1645G>A突变及c.IVS7-48T缺失,提示胎儿未患Sandhoff病(图2)。患儿母亲于妊娠39周自然分娩,新生女婴出生体重3 100 g,出生顺利,脐带血白细胞己糖胺酶A+B活性正常,HEXB基因分析未见异常,验证了产前诊断结果。
2 讨论
2.1 Sandhoff病临床表现 Sandhoff病临床表现与Tay-Sachs病相似,以进行性神经系统退行性病变为主要特征,个体差异较大,具有明显的临床异质,根据发病年龄及严重程度,分为3个表型,即婴儿型、青少年型及成人型[11]。本研究先证者临床表型符合Sandhoff病婴儿型。婴儿型病情较为严重,患儿在出生后数月内表现正常,偶见惊跳现象,常于4~6个月时出现惊厥、听觉过敏、视力减退及失明、肝脾肿大、肌张力减低,智力运动倒退,病情发展迅速,晚期出现去大脑强直及巨脑,大多数患儿在4岁前死亡。少数患儿伴随其它异常,如1例泰国患儿首先出现心脏二尖瓣脱垂伴回流,随后出现发育减退,惊跳,眼底樱桃红斑[12]。Venugopalan等[13]报道的1例婴儿患者除有二尖瓣脱垂伴反流外,还存在主动脉脱垂伴轻度反流及室间隔不均匀肥大。支气管和肺的发育异常[7]及双边并指畸形情况在Sandhoff病患者中也有报道[14-15]。
青少年型Sandhoff病一般在4~6岁起病,表现为痴呆、小脑共济失调、智力落后、肌肉萎缩,其严重程度仅次于婴儿型。Hendriksz等[16]报道了9例巴基斯坦血统的青少年患者,除共济失调外,还存在语言障碍、便秘、尿失禁以及下肢反射增强的症状;但眼底均未出现樱桃红斑改变。成人型Sandhoff病一般在儿童早期发病,至少有35%的患者在10岁前发病,临床表现进展缓慢[17]。Kohno 等[18]报道了日本首例成年Sandhoff病,患者于15岁发病,表现为运动神经元病变,首发症状为下肢近端无力。Ahn等[19]报道了1例23岁表现为运动神经元病变的成人,患者四肢无力,进行性运动障碍。Modigliani等[20]报道了1 例60 岁的Sandhoff病患者,临床表现为严重腹泻和轻微植物神经功能紊乱。Delnooz等[21]报道的6名晚发型患者均表现为小脑性共济失调。Pellegrini等[22]报道了2名慢性进行性Sandhoff病同胞,患者在60岁时出现进行性吞咽困难,同时伴有慢性运动神经疾病及自主神经受累。
2.2 Sandhoff病影像学特点 影像学检查是溶酶体病重要辅助诊断方法。一些Sandhoff病患者类似GM1神经节苷脂贮积症的骨骼改变[23]。本例患儿1岁7个月时骨龄骨质正常,但是脊柱后凸显著。Sandhoff病主要累及中枢神经系统,随疾病进展进行性加重,以丘脑、基底节(尾状核、壳核、苍白球)和小脑为著,一般很少累及其它部位[24-26]。由钙的堆积,典型Sandhoff病患者脑影像检查可见丘脑及基底节区异常信号特征性表现[27-28]。早在1993年,Caliskan等曾将CT双侧丘脑高密度影作为该病诊断性标志[29]。Wilken等报道的1名10个月Sandhoff病女患儿,头颅MRI显示脑室周围白质、锥体束、基底节、小脑半球异常信号[30]。Yüksel等[24]描述了 4 名患者,头颅 CT 显示双侧丘脑均匀高密度,MRI显示轻度皮质萎缩,胼胝体变薄,尾状核、苍白球、壳核、小脑和脑干异常信号。Yun等报道1例婴儿型患者脑MRIT2相示双侧丘脑低密度影、大脑白质高密度影,脑皮质及小脑萎缩[17]。本例患者1岁时脑MRI未见明显异常,1岁5个月来院时存在双侧基底节及弥漫性脑白质病变,符合疾病病理特点,提示脑影像学追踪观察对于神经变性病患者的病因诊断的重要性。
2.3 Sandhoff病的诊疗 与其他罕见的溶酶体病类似,Sandhoff病患者临床诊断困难,需要依赖酶学分析进行诊断和鉴别诊断[31],在明确HEXB基因突变的基础上,才能进行相关家系的遗传咨询及下一胎同胞的产前诊断[7]。
本例患儿诊断过程曲折,8个月后精神运动开始倒退,常有惊跳样反应,曾被诊断为脑性瘫痪,功能训练无效,病情进行性加重。于1岁2个月时全身瘫软,伴有脊柱后凸,于1岁4个月出现惊厥,并发现轻度肝肿大,考虑到遗传代谢病的可能,经血液氨基酸、酯酰肉碱谱分析及尿液有机酸分析排除了氨基酸、有机酸及脂肪酸代谢病。患儿1岁5个月来院就诊,眼底检查发现双侧樱桃红斑,结合病史中有惊跳、声音敏感、打鼾、痰多等异常,考虑到神经节脑苷脂病的可能性,通过己糖胺酶A+B活性分析及基因分析证实了Sandhoff病诊断。
眼底樱桃红斑是本病具有特征性的临床表现,由于黄斑周围神经节细胞内脂质沉积,至使该部位视网膜加厚失去透明度,而黄斑部本身不含神经节细胞,与周围之白晕相比而呈红色[17]。本例患儿1岁5个月就诊时双侧眼底均可见樱桃红斑,是获得病因诊断的关键线索之一。但并非所有患者眼底均出现此特征性改变,Hendriksz等报道的青少年型患者眼底未见樱桃红斑改变[16]。一些患者早期眼底检查正常,随着疾病的进展出现眼底改变及视力损害。因此,对于可疑患者需要随访,动态观察眼底病变。
迄今国内外已经报道了HEXB基因32种突变[7,32,34],本例患者存在第 14 外显子 1645G>A错义突变,为未报道的新突变,在100名正常对照及千人基因组计划数据库中均未检出(www.1000genomes.org)。c.1645G > A 突变导致己糖胺酶B的β肽链第549位氨基酸由甘氨酸改变为精氨酸。
Sandhoff病目前尚无特殊治疗方法,只能进行对症治疗。Tallaksen等[35]使用 Miglustat(zavesca)对一名挪威青少年患者进行治疗,并应用电休克疗法(ECT)缓解其抑郁症状,患者病情进展速度减慢,各项功能指标得以改善。也有研究将大脑内皮细胞作为治疗的靶向,诱导其分泌β-己糖胺酶[36],目前尚未获得有效证据。患者预后不良,多数婴儿型患者进行性衰竭,于4岁内夭折,死于惊厥、感染等合并症。
2.4 Sandhoff病的胎产前诊断 产前诊断是预防遗传性疾病再发的重要措施,在先证者病因诊断、基因型明确的基础上,在母亲下一次妊娠时可通过胎盘绒毛或羊水细胞的酶学或基因分析可进行Sandhoff病的产前诊断。Giles和Kaur等国外同行采用胎盘绒毛上皮细胞、羊水细胞酶学分析,成功地帮助一些Sandhoff病家系进行了产前诊断[37-38]。Warner采用高效液相法羊水N-乙酰葡糖胺寡糖进行Sandhoff病的产前诊断研究[39]。
本例患儿母亲于再次妊娠18周时来我院进行遗传咨询,希望对胎儿进行产前基因诊断。本研究首次采用羊水细胞HEXB基因分析为该家系的胎儿进行了产前诊断,证实胎儿未携带致病突变,出生后经脐带血白细胞酶学及基因分析进一步验证了产前诊断结果。
综上所述,Sandhoff病是一种罕见的GM2神经节苷脂沉积病,临床诊断困难,对于进行性神经系统退行性疾病的患者,应高度重视,通过生化检查、影像学检查逐步进行鉴别诊断。己糖胺酶A+B活性分析是疾病确诊的关键技术,基因分析则有助于遗传咨询及产前诊断。本文报告了一例婴儿型 Sandhoff病,明确了HEXB基因突变,并发现了一个新突变。首次采用羊水细胞基因分析方法,帮助该家系进行了下一胎的产前诊断。
[1]BLEY A E,GIANNIKOPOULOS O A,HAYDEN D,et al.Natural history of infantile G(M2)gangliosidosis[J].Pediatrics,2011,128(5):e1233-1241.
[2]SANDHOFF K,ANDREAE U,JATZKEWITZ H.Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs[J].Pathol Eur,1968,3(2):278-285.
[3]SAOUAB R,MAHI M,ABILKACEM R,et al.A case report of Sandhoff disease [J].Clin Neuroradiol,2011,21(2):83-85.
[4]MA Xiuwei,PU Lihua,ZHANG Yuehua,et al(马秀伟,蒲利华,张月华,等).Clinical characteristics and diagnosis of GM 2 gangliosidosis[J].Journal of Applied Clinical Pediatrics(实用儿科临床杂志),2008,23(7):539-541.(in Chinese)
[5]PROIA R L.Gene encoding the human betahexosaminidase beta chain:extensive homology of intron placement in the alpha-and beta-chain genes[J].Proc Nat Acad Sci,1988,85(6):1883-1887.
[6]HOU Lin,KOUSAKU O(侯 琳,Ohno KOUSAKU).Studies on the molecular mechanism of GM(2)gangliosidosis[J].Chinese Journal of Medical Genetics(中华医学遗传学杂志),2003,20(2):103-106.(in Chinese)
[7]FITTERER B B,ANTONISHYN N A,HALL P L,et al.A polymerase chain reaction-based genotyping assay for detecting a novel sandhoff disease-causing mutation[J].Genet Test Mol Biomarkers,2012,16(5):401-405.
[8]DEWJI N N,DE-KEYZER D R,STIRLING J L.Purification and characterization of β-N-acetylhexosaminid a se I 2 from human liver[J].Biochem J,1986,234(1):157-162.
[9]LIESSEM B,GLOMBITZA G J,KNOLL F,et al.Photoaffinity labeling of human lysosomal betahexosaminidase B.Identification of Glu-355 at the substrate binding site[J].J Biol Chem,1995,270(40):23693-23699.
[10]MILLER S A,DYKES D D,POLESKY H F.A simple salting out procedure for extracting DNA from human nucleated cells[J].Nucleic Acids Res,1988,16(3):1215.
[11]O'DOWD B F,KLAVINS M H,WILLARD H F,et al.Molecular heterogenicity in the infantile and juvenile forms of Sandhoff disease(O-variant GM2 gangliosidosis)[J].J Biol Chem,1985,261(27):12680-12685.
[12]SAKPICHAISAKUL K, TAERANAWICH P,NITIAPINYASAKUL A,et al.Identification of Sandhoff disease in a Thai family:clinical and biochemical characterization [J].J Med Assoc Thai,2010,93(9):1088-1092.
[13]VENUGOPALAN P, JOSHI S N. Cardiac involvement in infantile Sandhoff disease[J].J Paediatr Child Health,2002,38(1):98-100.
[14]ABDUL-WAHAB A,BESSISSO M S,ELSAID M F.Sandhoff disease(GM2 Gangliosidoses)in a premature patient with bronchopulmonary dysplasia[J].Saudi Med J,2002,23(5):602-605.
[15]NAIR P M,BATACLAN F,GANESH A.Sandhoff disease in an extreme preterm baby with bilateral syndactyly[J].Saudi Med J,2003,24(4):419-420.
[16]HENDRIKSZ C J,CORRY P C,WRAITH J E,et al.Juvenile Sandhoff disease-nine new cases and a review of the literature[J].J Inherit Metab Dis,2004,27(2):241-249.
[17]YUN Y M,LEE SN.A Case Refort of Sandhoff Disease[J].Korean J Ophthalmol,2005,19(1):68-72.
[18]KOHNO Y,YOSHIZAWA T,OHKOSHI N,et al.Adult Sandhoff disease presented as a motor neuron disease phenotype with slow progression[J].Rinsho Shinkeigaku,2001,41(1):36-39.
[19]AHN S W,KIM S H,HONG Y H,et al.A mutation of HEXB gene in Sandhoff disease presenting as motor neuron disease[J].Neurol India,2010,58(6):950-951.
[20]MODIGLIANI R,LEMANN M,MELAN ON S B,et al.Diarrhea and autonomic dysfunction in a patient with hexosaminidase B deficiency(Sandhoffdisease)[J].Gastroenterology,1994,106(3):775-781.
[21]DELNOOZ C C,LEFEBER D J,LANGEMEIJER S M,et al.New cases of adult-onset Sandhoff disease with a cerebellar or lower motor neuron phenotype[J].J Neurol Neurosurg Psychiatry,2010,81(9):968-972.
[22]PELLEGRINI M,ZICARI E,DOTTI M T,et al.Dysautonomic achalasia in two siblings with Sandhoff disease[J].J Neurol Sci,2006,241(1-2):107-109.
[23]SHI Huiping(施 惠 平).Lysosomal Storage Disorders[J].Journal of Applied Clinical Pediatrics(实用儿科临床杂志),2007,22(8):561-563.(in Chinese)
[24]YÜKSEL A,YALÇINKAYA C,I爦LAK C,et al.Neuroimaging findings of four patients with Sandhoff disease [J].Pediatr Neurol,1999,21(2):562-565.
[25]GROSSO S,FARNETANI MA,BERARDI R,et al.GM2 gangliosidosis variant B1 neuroradiological findings[J].J Neurol,2003,250(1):17-21.
[26]BANO S,PRASAD A,YADAV S N,et al.Neuroradiological findings in GM2 gangliosidosis variant B1 [J].J Pediatr Neurosci,2011,6(2):110-113.
[27]GIRISHA K M,PHADKE S R.Basal ganglia changes:a diagnostic clue to Sandhoff disease[J].Indian Pediatr,2006,43(10):919-920.
[28]HITTMAIR K,WIMBERGER D,BERNERT G,et al.MRI in a case of Sanhoff's disease [J].Neuroradiology,1996,38(Suppl 1):S178-180.
[29]CALISKAN M,OZMEN M,BECK M,et al.Thalamic hyperdensity-is it a diagnostic marker for Sandhoff disease?[J].Brain Dev,1993,15(5):387-388.
[30]WILKEN B,DECHENT P,HANEFELD F,et al.Proton MRS of a child with Sandhoff disease reveals elevated brain hexosamine[J].Eur J Paediatr Neurol,2008,12(1):56-60.
[31]CHAMOLES N A,BLANCO M,GAGGIOLI D,et al.Tay-Sachs and Sandhoff diseases:enzymatic diagnosis in dried blood spots on filter paper:retrospective diagnoses in newborn-screening cards[J].Clin Chim Acta,2002,318(1-2):133-137.
[32]ZAMPIERI S,FILOCAMO M,BURATTI E,et al.Molecular and functional analysis of the HEXB gene in Italian patients affected with Sandhoff disease:identification of six novel alleles[J].Neurogenetics,2009,10(1):49-58.
[33]YOSHIZAWA T,KOHNO Y,NISSATO S,et al.Compound heterozygosity with two novel mutations in the HEXB gene produces adult Sandhoff disease presenting as a motor neuron disease phenotype[J].J Neurol Sci,2002,195(2):129-138.
[34]WANG S Z,CACH N-GONZ LEZ M B,STEIN P E,et al.A novel HEXB mutation and its structural effects in juvenile Sandhoff disease [J].Mol Genet Metab,2008,95(4):236-238.
[35]TALLAKSEN C M,BERG J E.Miglustat therapy in juvenile Sandhoff disease[J].J Inherit MetabDis,2009,32(Suppl 1):S289-293.
[36]BATISTA L,MILLER F,CLAVE C,et al.Induced secretion of beta-hexosaminidase by human brain endothelial cells:a novel approach inSandhoff disease?[J].Neurobiol Dis,2010,37(3):656-660.
[37]GILES L,COOPER A,FOWLER B,et al.First trimester prenatal diagnosis of Sandhoff's disease[J].Prenat Diagn,1988,8(3):199-205.
[38]KAUR M,VERMA I C.Enzyme studies in GM2 gangliosidiosis,and their application in prenatal diagnosis[J].Indian J Pediatr,1995,62(4):485-489.
[39]WARNER T G,TURNER M W,TOONE J R,et al.Prenatal diagnosis of infantile GM 2 gangliosidosis type II(Sandhoff disease)by detection of N-acetylglucosaminyl-oligosaccharides in amniotic fluid with high-performance liquid chromatography[J].Prenat Diagn,1986,6(6):393-400.
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
心电与循环(2021年4期)2021-11-29
健康之家(2021年19期)2021-05-23
锻压装备与制造技术(2020年6期)2021-01-25
科技资讯(2020年32期)2020-12-28
宝藏(2020年3期)2020-10-14
中国现代神经疾病杂志(2020年9期)2020-01-09
中国心血管杂志(2020年4期)2020-01-09
中医眼耳鼻喉杂志(2019年2期)2019-04-13
中医眼耳鼻喉杂志(2019年2期)2019-04-13
