
重组a-氨基酸酯水解酶合成头孢曲嗪
2013-11-12 02:20:38潘佳林王辂李端华叶丽娟
生物工程学报 2013年4期
潘佳林,王辂,李端华,叶丽娟
中国医药集团总公司四川抗菌素工业研究所,四川 成都 610052
头孢曲嗪为半合成的广谱口服头孢菌素,对b-内酰胺酶具有很高的稳定性[1]。用于制备头孢曲嗪的原料成本低于头孢克洛,临床疗效与头孢克洛类似[2]。因此,头孢曲嗪可作为头孢克洛的替代品种,市场潜力巨大。目前头孢曲嗪的制备方法主要为化学方法,一方面残留的溶剂威胁患者的用药安全,另一方面产生的废弃溶媒造成一定的环保压力。室温条件下和在中性水溶液中进行的酶催化合成具有更高的能效和更低的碳排放量[3],利用酶法合成路线代替化学合成路线可以产生巨大的环境和社会效益[4-5]。
虽然近年来酶法合成头孢类抗生素的研究比较广泛[6-8],但目前还没有关于头孢曲嗪酶法合成工艺的文献报道。a-氨基酸酯水解酶(a-Amino acid ester hydrolase,简称 AEH,EC 3.1.1.43) 是一类具备头孢曲嗪合成活性的酶。1972年由Takahashi等在寻找能够合成带a-氨基的抗生素的过程中被发现[9]。由于底物范围限于带a-氨基的酰基供体,而且相比酰胺,对酯的亲和性更高而得名[10]。AEH同时催化3个反应 (图1):1)催化酰胺键的形成,酶发挥抗生素合成酶活性;2)催化酰基供体的水解;3)催化产物的水解,酶发挥酰胺酶活性[11]。酶法合成头孢类抗生素可以通过热力学控制 (Thermodynamically controlled synthesis,TCS)[12]或者动力学控制(Kinetically controlled synthesis,KCS)[13]来实现。与热力学合成不同,动力学控制最终的转化率不是由平衡状态决定,而是由3个反应的速率共同决定[14]。动力学控制的反应其特点是存在暂时的最大转化率[15]。由于产物浓度不再受反应平衡的限制,对于高转化率意味着低成本的制药工业来说,动力学无疑是比热力学更好的策略。本文从AEH的异源表达开始,采用纯化的重组酶催化合成头孢曲嗪,以动力学控制策略考察了特定条件下头孢曲嗪的转化率。
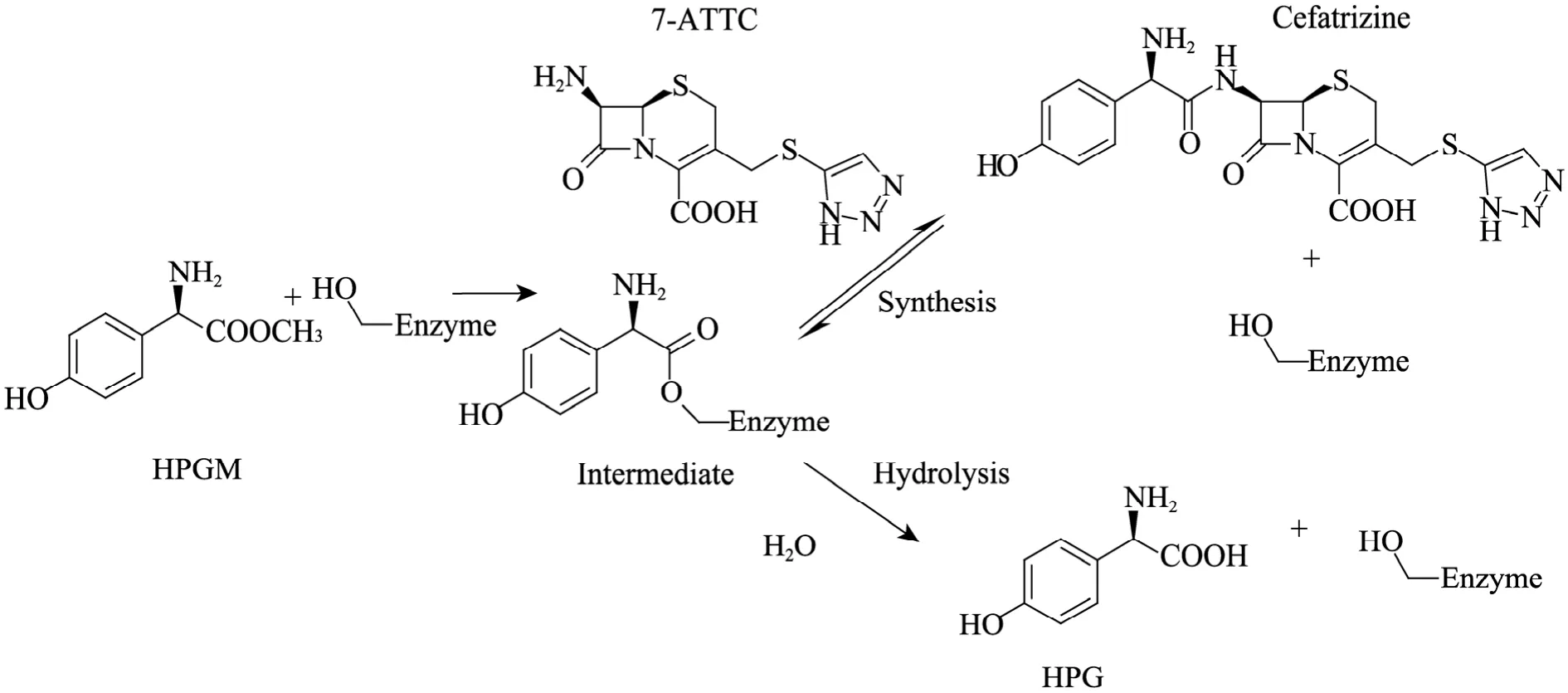
图1 AEH催化的头孢曲嗪的合成Fig. 1 Synthesis of cefatrizine by AEH.
1 材料与方法
1.1 菌株与质粒
红纹黄单胞菌 Xanthomonas rubrillineans CPCC 140817由中国医药集团四川抗菌素工业研究所保藏;E. coli BL21 (DE3)感受态细胞购自北京天根生化公司。克隆载体pGEM-T、表达载体pET28分别为Promega公司和Invitrogen公司产品。
1.2 引物与试剂
引物合成与测序由上海 Invitrogen公司完成。用于克隆的聚合酶为上海生工生物工程公司Pfu聚合酶,限制性内切酶EcoRⅠ和XhoⅠ购自Fermentas公司,质粒小量提取采用Bio-Tek公司的OMEGA试剂盒。BCA蛋白含量测定试剂盒为Pierce公司产品。亲和层析柱 (FF crude,5 mL)购自GE Healthcare公司。超滤离心管 (Amicon,Ultra-15,50 kDa)为 Millipore公司产品。Sephadex G-200购自Pharmacia公司。
头孢氨苄标准品购自石家庄华北制药集团。7-氨基去乙酰氧基头孢烷酸 (7-ADCA)、苯苷氨酸甲酯盐酸盐 (PGM·HCl)、对羟基苯苷氨酸甲酯盐酸盐 (HPGM·HCl)为上海邦成化工有限公司产品,7-氨基-3-(1, 2, 3-三唑-4-硫基)甲基头孢烷酸 (7-ATTC)及头孢曲嗪标准品由四川抗菌素工业研究所化学部提供 (纯度≥98%)。其他试剂为进口或国产分析纯。
1.3 方法
1.3.1 aeh基因的克隆及转化大肠杆菌
采用 PCR扩增方法从红纹黄单胞菌X. rubrillineans CPCC 140817的全基因组中分离aeh基因。引物序列如下: 正向: 5'-CGGAATTC A TGCGCCGCATCGCTCCCTGCCTGC-3', 反向:5'-CCGCTCGAGTCAATGTACCGGCAGCTGAT GAAAC-3' (下划线为限制性酶切位点)。PCR反应体系:基因组DNA约100 ng,引物10 pmol/L,dNTPs 200 pmol/L,1×PCR缓冲液,Taq酶 1.5 U,ddH2O补足总体系至25 μL。梯度PCR反应程序如下:94 ℃ 3 min;98 ℃ 30 s,50~58 ℃ 1 min,72 ℃ 1.5 min,20 个循环;72 ℃,10 min。将 PCR产物克隆到pGEM-T载体中,测序验证后,再将pGEM-T-aeh和 pET28载体经EcoRⅠ和 XhoⅠ酶切后回收,相连并转化感受态大肠杆菌BL21 (DE3)。
1.3.2 重组AEH的酶活测定
以头孢氨苄合成活性跟踪 AEH的纯化过程。头孢氨苄合成活性测定方法如下:30 mmol/L 7-ADCA、15 mmol/L PGM·HCl溶于 50 mmol/L pH 6.2的磷酸钠缓冲液,酶的加量为50 μL酶溶液/(mL反应混和物),30 ℃下温育30 min。1 min生成1 μmol头孢氨苄所需的酶量定义为1个活力单位 (1 U)。HPLC 法测定反应生成头孢氨苄的量[16]。
1.3.3 重组AEH的表达及分离纯化
在10 L发酵罐中培养重组大肠杆菌。培养液装量6 L,培养液组成:乳糖1 g/L,胰蛋白胨10 g/L,酵母提取物5 g/L,NaCl 10 g/L,卡那霉素30 mg/L,聚醚消泡剂1 mL/L。接种量1%。发酵条件:温度25 ℃;通气量9 L/min;搅拌转速250 r/min。发酵周期24 h。
发酵结束后,冷冻离心机5 000×g离心收集菌体。参考Sambrook的方法[17]制备无细胞提取物。表达的蛋白N-端带有6×His标签,选择亲和层析纯化无细胞提取物。6个柱体积的结合缓冲液 (20 mmol/L磷酸钠,500 mmol/L NaCl,pH 7.8)洗柱后,以5 mL/min流速上样。依次用洗涤缓冲液 (10 mmol/L咪唑,20 mmol/L磷酸钠,500 mmol/L NaCl,pH 7.8)、洗脱缓冲液(50 mmol/L咪唑,20 mmol/L磷酸钠,500 mmol/L NaCl,pH 7.8)洗柱,分布收集洗脱液。最后用含有更高浓度咪唑的缓冲液洗柱 (200 mmol/L咪唑,20 mmol/L磷酸钠,500 mmol/L NaCl,pH 7.8)。经过SDS-PAGE分析,合并目标蛋白纯度较高的收集管,超滤离心管去除小分子盐并浓缩酶溶液。
分离纯化每一步获得的酶液按 1.3.2项下测量酶活。Pierce BCA蛋白含量测定试剂盒测定蛋白含量。
1.3.4 重组AEH亚基分子量的测定
变性聚丙烯酰胺凝胶电泳法测定亚基分子量。SDS-PAGE分析条件:10%分离胶,5%浓缩胶,上样量15 μL。采用Bio-Rad公司Powerpac Basic电泳系统,分子量标准为 Fermentas PageRuler Prestained Protein Ladder,考马斯亮蓝染色。
1.3.5 重组AEH合成头孢曲嗪最适反应pH与最适反应温度的确定
采用不同pH值的缓冲体系,分别测定重组AEH合成头孢曲嗪的反应初速度,确定重组AEH催化头孢曲嗪合成的最适pH值。30 mmol/L 7-ATTC、15 mmol/L HPGM×HCl溶于 50 mmol/L不同pH的缓冲液。pH 5.5~6.0,采用Na2HPO4-柠檬酸缓冲液;pH 6.5~7.5,采用 Na2HPO4-NaH2PO4缓冲液。酶加量为50 μL酶溶液/(mL反应混和物),30 ℃下温育30 min。测定生成的头孢曲嗪的量。
在最适 pH条件下,测定不同温度下重组AEH催化头孢曲嗪合成的反应过程。30 mmol/L 7-ATTC、15 mmol/L HPGM×HCl溶于 50 mmol/L Na2HPO4-柠檬酸缓冲液 (pH 6.0),酶加量为50 μL酶溶液/(mL反应混和物),分别在28 ℃、32 ℃、36 ℃、40 ℃下温育180 min,测定生成的头孢曲嗪的量。
1.3.6 两底物最佳摩尔比的确定
底物溶液配置:以初始反应体积100 mL计算所需2种底物的量。0.3 mol/L的磷酸钠缓冲液(pH 7.5)溶解0.94 g 7-ATTC (30 mmol/L),缓慢滴加少量1 mol/L NaOH帮助溶解,pH计监控溶解过程pH不超过8.0。缓慢加入HPGM×HCl,搅拌溶解,0.3 mol/L的磷酸钠缓冲液 (pH 6.2)定容至 96 mL (加入酶溶液后初始反应体系约为100 mL)。用1 mol/L NaOH调整pH至6.0。
催化反应:向底物混合物中加入酶液开启反应,酶加量为 22 U/(mL反应体系)。酶反应在36 ℃,pH为(6.0±0.1)的条件下进行,以pH自动控制器控制pH并自动流加1 mol/L NaOH,记录所加NaOH的体积,用于计算即时反应体积。取样初始反应混和物,HPLC测定2种底物的初始浓度。反应过程中间隔取样,HPLC分析反应体系中各组分浓度。
1.3.7 HPLC检测条件
去离子水稀释反应体系中取出的样品,使浓度介于各组分标准曲线线性范围内。10 000×g离心后,HPLC分析上清液。分析柱:Ultimate XB-18(200 mm×46 mm,5 μm)。流动相:A 相:MeOH;B相:50 mmol/L磷酸铵缓冲液 (pH 2.1),A∶B=15∶85 (V/V)。B相配置方法:3 mL 85%磷酸溶于950 mL去离子水,25%氨水溶液调整pH至2.1,去离子水定容至 1 L后混匀备用。流速:1 mL/min,柱温箱:35 ℃。检测波长:230 nm。
2 结果
2.1 重组AEH的克隆、纯化与鉴定
PCR扩增得到与预期大小一致的条带 (约2 kb)。经回收测序分析,确定为aeh基因,提交GenBank获得登录号JF744990。
亲和层析步骤中,含50 mmol/L咪唑的洗脱缓冲液可以将绝大部分AEH洗脱下来。经过超滤离心浓缩后的酶溶液经SDS-PAGE分析,重组蛋白单亚基的分子量约为 72 kDa,与报道的X. rubrillineans AEH的单亚基分子量一致[18](图 2)。
大肠杆菌BL21(DE3)表达的重组AEH大约占无细胞提取物中总蛋白的1%。经过3个步骤的提取,从 1 L大肠杆菌的发酵液中可获得约1.7 mg 纯度为90%的AEH (表1)。
2.2 重组AEH催化合成头孢曲嗪最适pH和温度
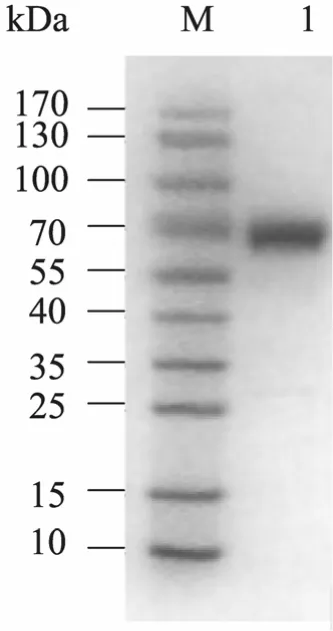
图2 经纯化的重组AEH电泳图Fig. 2 SDS-PAGE of purified recombinant AEH. M:protein marker; 1: sample of recombinant AEH after affinity chromatography.

表1 大肠杆菌BL21(DE3)中的重组AEH的纯化Table 1 Purification of recombinant AEH from E. coli BL21(DE3)
分离自X. rubrillineans的AEH其稳定pH范围介于5.0~8.0[19]。因此将重组AEH催化头孢曲嗪合成的pH考察范围定在5.5~7.5。如图3所示,pH 6.0的缓冲体系中重组AEH催化头孢曲嗪合成的初速度最高。
考虑到底物溶解度[20]和酶的温度稳定性[21],将合成反应温度的考察范围定在28 ℃~40 ℃之间。在 pH为 (6.0±0.1)的条件下,不同温度下重组AEH合成头孢曲嗪的反应过程如图4所示,最适温度下生成的头孢曲嗪的量标准化为100%。反应初速度随温度升高而增加,温度越高,达到最高反应转化率的时间越短。以反应转化率为指标,重组AEH头孢曲嗪合成的最佳温度为36 ℃。反应温度的选择需要综合考虑几个方面的因素,温度不仅影响酶催化的速度,也会影响反应各组分溶解性及反应产物的稳定性。相对较低的温度可以增加酶及产物稳定性,但是达到最高转化率的时间可能延长而导致工业化之后的时间成本增加。
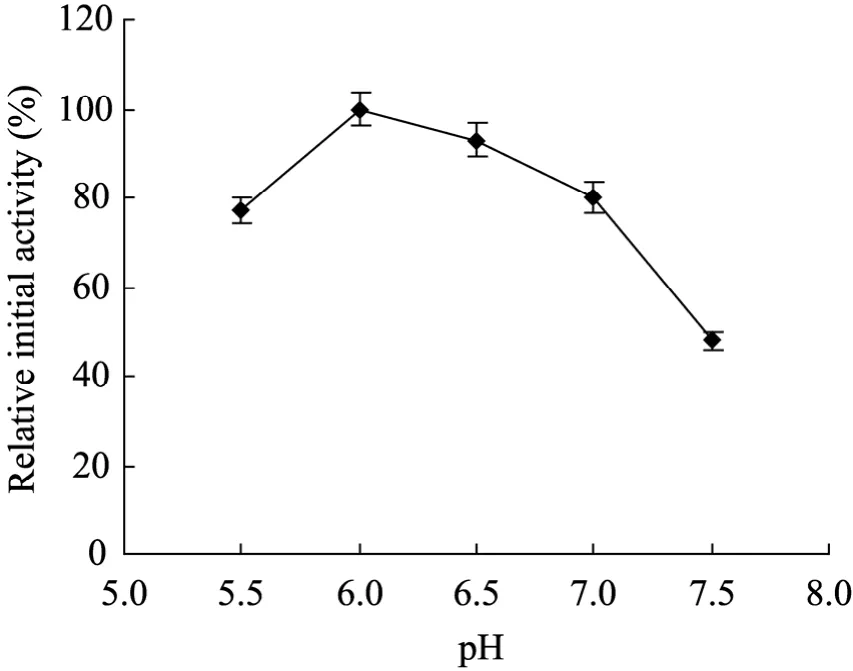
图3 pH对AEH合成头孢曲嗪初速度的影响Fig. 3 Effect of pH on initial activity of AEH toward cefatrizine synthesis. The initial activity under optimal pH was standardized to 100%. All synthesis experiments were performed in triplicate by purified enzyme.
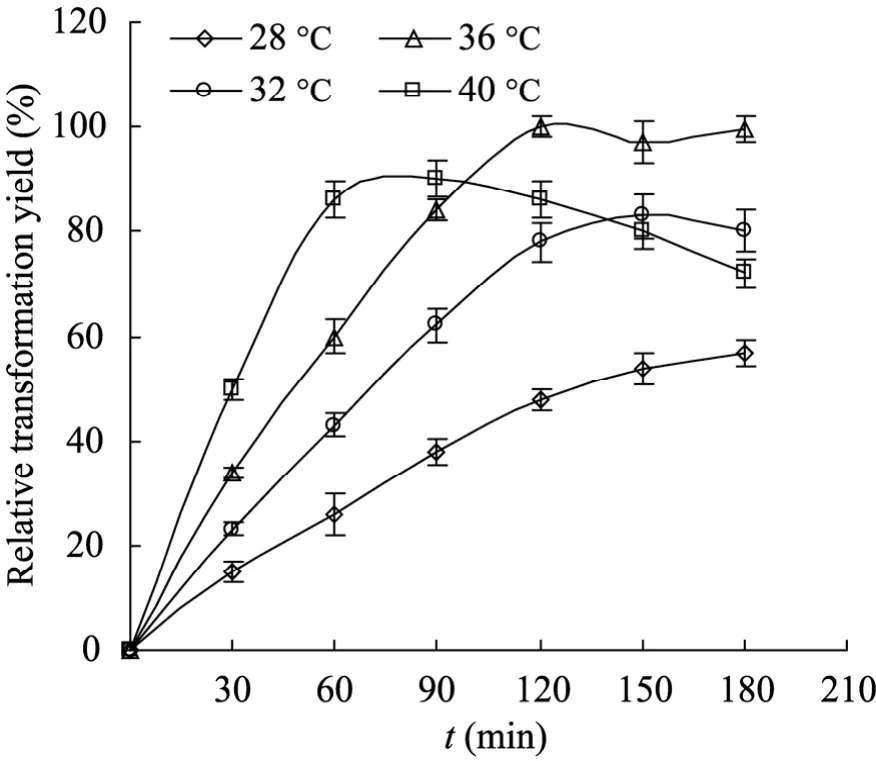
图4 温度对AEH合成头孢曲嗪初速度的影响Fig. 4 Effect of temperature on initial activity of AEH toward cefatrizine synthesis. The initial activity under optimal temperature was standardized to 100%. All synthesis experiments were performed in triplicate by purified enzyme.
2.3 两底物不同摩尔比条件下头孢曲嗪的合成
反应温度36 ℃,反应pH为 (6.0±0.1)的条件下,当母核7-ATTC的浓度约为30 mmol/L,侧链HPGM×HCl与母核初始摩尔比分别为2.0、2.9和4.1时,头孢曲嗪的酶法合成实验结果见表2。合成转化率ηCFTZ随着摩尔比的增加而增加。
侧链与母核摩尔比为4.1的条件下,整个反应过程中底物的减少以及产物的增加见图5。转化率在150 min到达峰值,后续90 min内,头孢曲嗪的浓度基本不变。从工业角度看该现象非常有利,有足够的时间终止酶催化反应以及进行下游过程处理,不必担心产物的水解。合成转化率最高处,HPGM大约剩余40%,此后的HPGM没有用于合成头孢曲嗪,而是缓慢水解为HPG。
图5也显示了相对于母核和侧链浓度的物质平衡情况。整个催化反应过程中,含HPG化合物 (HPG+HPGM+头孢曲嗪)的物质平衡非常好,介于 97%~101%之间,含母核化合物(7-ATTC+头孢曲嗪)的物质平衡随着反应的进行逐渐下降,在最高转化率处,即150 min时为89.9%,至反应终止时为81.7%。
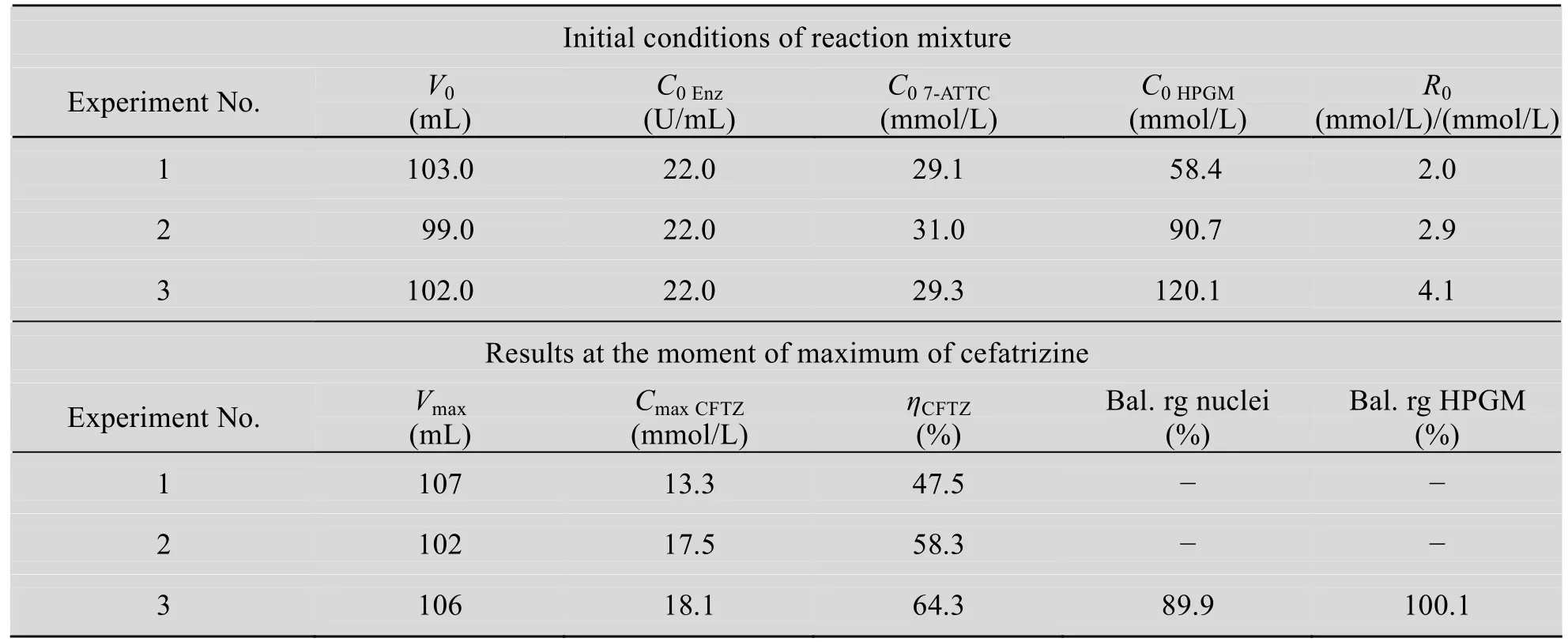
表2 重组AEH催化7-ATTC和HPGM合成头孢曲嗪Table 2 Enzymatic synthesis of cefatrizine from 7-ATTC and HPGM by AEH
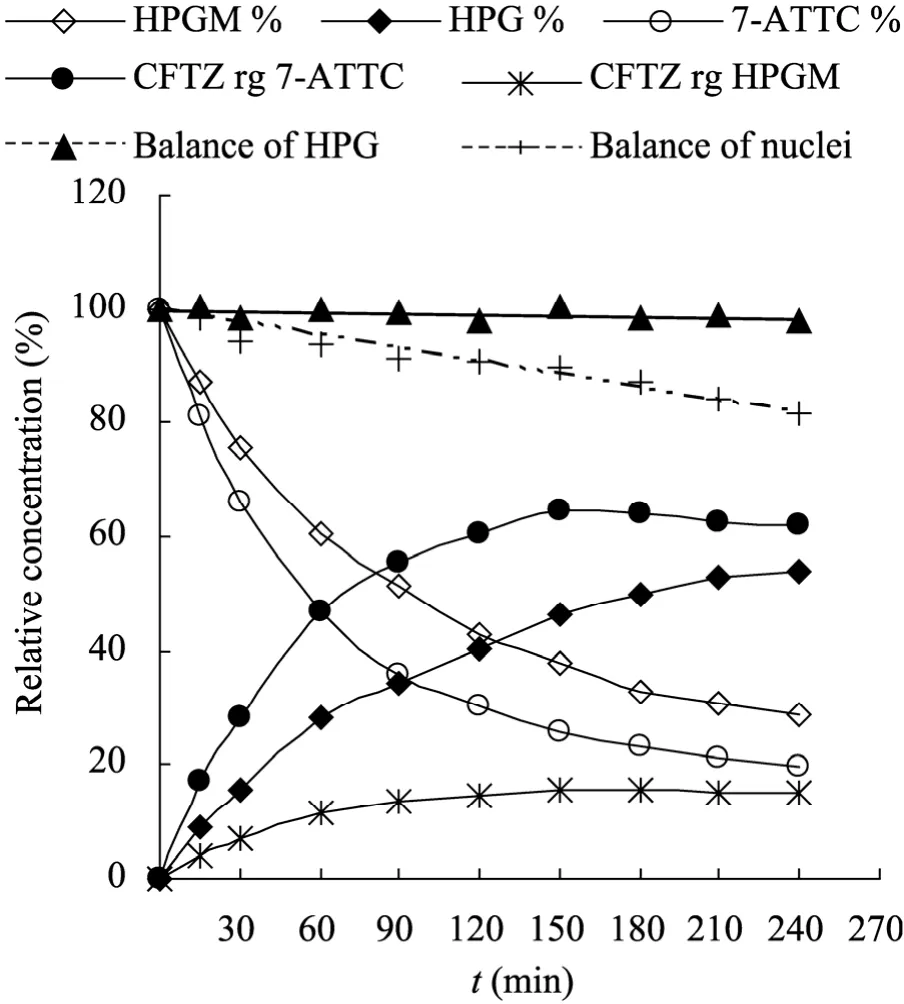
图5 AEH催化的头孢曲嗪合成动态过程Fig. 5 Dynamics of cefatrizine synthesis by AEH.Initial masses of 7-ATTC and HPGM were standardized to 100%.
3 讨论
由于实验是产业化工艺的前期研究,为了保障工艺规模放大后的重现性,每一次头孢曲嗪酶法合成实验都采用 HPLC精确测定底物初始浓度,避免了通过称量和定容引起的误差。反应过程定量调节pH导致的反应体积增加,保证对最高转化率时间点处各组分的精确定量。
母核物质平衡在整个反应过程中呈现逐步下降的趋势,可能与母核7-ATTC的某种副反应有关。在所有反应混合物样品的HPLC图谱中出现了与4个标准品不一致的2个未知的峰 (结果未显示)。可以确定的是该副反应与酶相关,因为以缓冲液代替酶液的反应空白对照并没出现未知峰,具体是何物质有待进行收集和解析,以探索排除该副反应的可能性。
虽然形成头孢曲嗪需要的酰基受体 (母核7-ATTC)和供体 (HPGM)的摩尔比相等,由于HPGM存在水解副反应,HPGM的过量可以提高产物转化率。这也是通过动力学控制使反应向合成方向进行的体现。头孢氨苄、头孢唑啉和头孢克洛也有采用该策略的报道[22]。虽然侧链成本低于母核,但显然无限制升高侧链浓度并不是提高转化率的明智选择。因此实验选择的摩尔比范围介于2∶1~4∶1。下一步将在此基础上尝试添加有机溶剂抑制水解或采用产物络合的方法提高合成转化率。
通过本实验一方面实现了 AEH的异源表达,为降低酶的生产成本提供了可能;另一方面,确定了反应体系中各成分的 HPLC分析方法和物质量衡算方法。不过,头孢曲嗪64.3%的转化率距离实现产业化还有很长的路要走,可以选择很多策略来改善转化率,比如:通过分子改造[23]或酶制剂形式[24]提高催化合成头孢曲嗪的效率,或通过改变反应体系使反应平衡倾向合成反应[25]等等。
[1]Essack SY. The development of β-lactam antibiotics in response to the evolution of β-lactamases. Pharm Res, 2001, 18(10):1391−1399.
[2]Fung-Tomc JC, Huczko E, Stickle T, et al.Antibacterial activities of cefprozil compared with those of 13 oral cephems and 3 macrolides.Antimicrob Agents Chemother, 1995, 39(2):533−538.
[3]Woodley JM. New opportunities for biocatalysis:making pharmaceutical processes greener. Trends Biotechnol, 2008, 26(6): 321−327.
[4]Bruggink A, Roy PD. Industrial synthesis of semi-synthetic antibiotics//Bruggink A. Synthesis of β-lactam Antibiotics, Chemistry, Biocatalysis and Process Integration. Dordrecht: Kluwer Academic Publishers, 2001: 13−56.
[5]Volpato G, Rodrigues RC, Fernandez-Lafuente R.Use of enzymes in the production of semi-synthetic penicillins and cephalosporins: drawbacks and perspectives. Curr Med Chem, 2010, 17(32):3855−3873.
[6]Valencia P, Flores S, Wilson L, et al. Batch reactor performance for the enzymatic synthesis of cephalexin: influence of catalyst enzyme loading and particle size. New Biotechnol, 2012, 29(2):218−226.
[7]Sheldon RA. Characteristic features and biotechnological applications of cross-linked enzyme aggregates (CLEAs). Appl Microbiol Biotechnol, 2011, 92(3): 467−477.
[8]Bahamondes C, Wilson L, Aguirre C, et al.Comparative study of the enzymatic synthesis of cephalexin at high substrate concentration in aqueous and organic media using statistical model.Biotechnol Bioproc E, 2012, 17(4): 711−721.
[9]Takahashi T, Yamazaki Y, Kato K, et al. Enzymic synthesis of cephalosporins. J Am Chem Soc, 1972,94(11): 4035−4037.
[10]Blum JK, Bommarius AS. Amino ester hydrolase from Xanthomonas campestris pv. campestris,ATTC 33913 for enzymatic synthesis of ampicillin.J Mol Catal B Enzym, 2012, 67(1/2): 21−28.
[11]Barends TRM, Polderman-Tijmes JJ, Jekel PA, et al. The sequence and crystal structure of thea-amino acid ester hydrolase from Xanthomonas citri define a new family of b-lactam antibiotic acylases. J Biol Chem, 2003, 278(25):23076−23084.
[12]Schroën CGPH, Nierstrasz VA, Kroon PJ, et al.Thermodynamically controlled synthesis of β-lactam antibiotics. Equilibrium concentrations and side-chain properties. Enzyme Microb Tech,1999, 24(8/9): 489−506.
[13]Schroën CGPH, Nierstrasz VA, Moody HM, et al.Modeling of the enzymatic kinetic synthesis of cephalexin-influence of substrate concentration and temperature. Biotechnol Bioeng, 2001, 73(3):171−178.
[14]Diender MB, Straathof AJJ, Heijnen JJ. Predicting enzyme catalyzed reaction equilibria in cosolvent-water mixtures as a function of pH and solvent composition. Biocatal Biotransform, 1998,16(4): 275−289.
[15]Polderman-Tijmes JJ. Biochemical characterization of a-amino acid ester hydrolases[D]. Groningen:University of Groningen, 2004.
[16]Xia DN, Xia YJ, Yin J, et al. Simultaneous determination of cefalexin and trimethoprim in plasma by HPLC. Chin J Antibiot, 2008, 33(11):682−684, 700 (in Chinese).夏登宁, 夏艳姣, 尹佳, 等. HPLC同时测定血浆中头孢氨苄和甲氧苄啶的浓度. 中国抗生素杂志, 2008, 33(11): 682−684, 700.
[17]Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed. Beijing: Science Press,2002: 1252 (in Chinese).萨姆布鲁克 J, 拉塞尔 DW. 分子克隆实验指南.3版. 北京: 科学出版社, 2002: 1252.
[18]Krest'ianova IN, Uvarov NN, Rudenskaia GN, et al.Intracellular aminopeptidase from Xanthomonas rubrilineans, hydrolyzing a-amino acid esters and cefalexin. Biokhimiia, 1990, 55(12): 2226−2238.
[19]Qu F, Yi BX, Ye LJ. Purification and characterization of a-amino acid ester hydrolase from Xanthomonas rubrillineans. Acta Microbiol Sin, 2012, 52(5): 620−628 (in Chinese).屈凤, 易八贤, 叶丽娟. 红纹黄单胞菌a-氨基酸酯水解酶的分离纯化及酶学性质. 微生物学报,2012, 52(5): 620−628.
[20]Kurochkina VB, Sklyarenko AV, Satarov JE, et al.Ionization constants and solubility of compounds involved in enzymatic synthesis of aminopenicillins and aminocephalosporins. Bioproc Biosyst Eng,2011, 34(9): 1103−1117.
[21]Bluma JK, Rickettsc MD, Bommarius AS.Improved thermostability of AEH by combining B-FIT analysis and structure-guided consensus method. J Biotechnol, 2012, 160(3/4): 214−221.
[22]Nys PS, Kurochkina VB. Methodological approach to development of enzymatic technologies for semisynthetic betalactam antibiotic production.Appl Biochem Biotech, 2000, 88(1/3): 221−229.
[23]Bornscheuer UT, Huisman GW, Kazlauskas RJ, et al. Engineering the third wave of biocatalysis.Nature, 2012, 485(7397): 185−194.
[24]Cecchini DA, Pavesi R, Sanna S, et al. Efficient biocatalyst for large-scale synthesis of cephalosporins, obtained by combining immobilization and site-directed mutagenesis of penicillin acylase. Appl Microbiol Biotechnol,2012, 95(6): 1491−1500.
[25]Feng SX, Liang SZ, Lou WY. Two-step, one-pot enzymatic synthesis of cefprozil from-phenylacetamido-3-propenyl-cephalosporanic acid(GPRA). Biocatal Biotransform, 2008, 26(4):321−326.
猜你喜欢
云南化工(2021年6期)2021-12-21 07:30:56
中国科技纵横(2021年24期)2021-03-02 06:42:52
科学(2020年2期)2020-08-24 07:57:00
中成药(2018年6期)2018-07-11 03:01:12
乐活老年(2016年10期)2016-02-28 09:30:32
生物技术通报(2015年1期)2015-04-10 16:15:19
中国药业(2014年12期)2014-06-06 02:17:26
中国合理用药探索(2014年11期)2014-03-11 20:30:22
当代畜禽养殖业(2014年4期)2014-02-27 07:58:54
山西大同大学学报(自然科学版)(2013年5期)2013-09-13 10:44:14
