
构建定向T载体用于基因克隆和表达
2013-11-12 02:20:38钟星翟超陈亮余晓岚蒋思婧严红杨登想马立新
生物工程学报 2013年4期
钟星,翟超,陈亮,余晓岚,蒋思婧,严红,杨登想,马立新
1 湖北大学生命科学学院,湖北 武汉 430062
2 湖北省工业生物技术重点实验室,湖北 武汉 430062
3 湖北大学知行学院生物工程系,湖北 武汉 430011
随着越来越多的物种基因组测序工作被完成,基因组数据库收录的基因组信息日益丰富,由已知序列设计引物扩增DNA并克隆,再进行相关结构、功能的研究,加快了生命科学研究的进程[1-2]。聚合酶链反应 (PCR)是最强有力的基因克隆方法,为了直接克隆PCR产物,T载体被广泛使用[3-5]。尤其当面对大量的克隆工作时,使用T载体克隆PCR产物便成为最经济简便的做法。传统的 T载体克隆方法利用 LacZ alpha插入失活筛选插入事件[6],但是由于 LacZ基因的移码、载体自身发生异常重组等事件将导致产生一部分假阳性白色菌落,转化后的白色菌落仍然需要被大量培养进行质粒抽提和鉴定[7-8];利用传统的T载体进行克隆表达时,由于插入片段的方向具有随机性,无法保证定向克隆,仍然需要筛选定向插入的克隆。
为了克服传统 T载体克隆方法需要大量鉴定重组子和无法保证定向克隆的缺点,我们设计了一个 LacZ正向筛选方案来实现免鉴定重组子、定向T载体克隆基因的方法。如图1B所示,在非诱导条件下的Lac+的宿主菌中,Lac启动子下游LacO序列被LacO结合蛋白LacI所阻遏,LacZ基因的表达处于抑制状态,该菌落在X-gal底物平板上显示为白色;如果基于pBR322的高拷贝载体上含有 LacO序列,该载体将从 LacZ启动子单元上竞争性吸附宿主菌内源LacI蛋白,对宿主菌的乳糖启动子去抑制[9-10],表达β-半乳糖苷酶并降解X-gal底物,使该菌落显示为蓝色(图 1C)。pET-23a(+)是基于 pBR322改造的高拷贝载体。我们在pET-23a(+)的基础上构建了定向T载体pETG,利用PCR片段和定向T载体连接重构完整的LacO序列 (图1A),转化Lac+菌株,使含有正确克隆的菌落变蓝来指示定向插入的重组事件 (图 1C)。对转化后平板上的蓝色菌落接种抽提质粒后酶切和 PCR鉴定表明蓝色菌落全部为定向插入的重组子。本研究利用构建的定向T载体成功地克隆了103个人类肝蛋白编码基因,随机挑选其中 8个基因的克隆进行蛋白质表达,均获得成功表达。对比传统方法,利用该T载体进行基因克隆非常简单、高效、可靠,并且可以实现基因定向插入,实验过程无需鉴定重组子,避免了大量抽提质粒和PCR鉴定的步骤,节约了大量的时间和试剂成本,同时该定向T载体还兼容传统T载体基因克隆,该定向 T载体有大规模高通量的基因克隆和表达的应用前景。
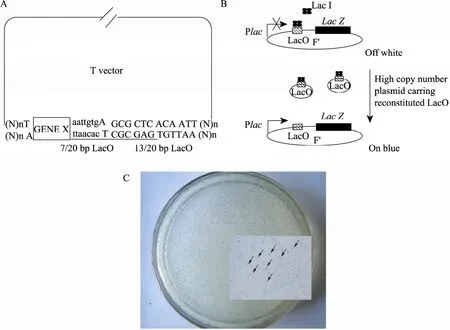
图1 定向T载体克隆原理Fig. 1 Principle of a directional T vector cloning method. After ligation, the insert and vector will reconstituted a full length of ideal LacO(A); reconstituted full length of ideal LacO occurring on a high copy number plasmid pETG will titrate out of the lac repressors (LacI )which adhered to the LacO sites on F` plasmid, thus LacZ transcription is derepressed (B), and the colonies go blue (C). Followed by transformation, the recombinant events were phenotypically selected, transformants with blue color revealed the desired recombinants.
1 材料与方法
1.1 菌株和质粒
表达载体pET-23a(+)、用于表达的大肠杆菌BL21(DE3)购自Novagen公司,用于克隆的大肠杆菌XL10-Gold购自Stratagen公司,DH10β [基因型:mcrAΔ (mrr-hsdRMS-mcrBC)ф80lacZM15 ΔlacX74deoR recA1 araΔ 139 (ara,leu)7697 galU galK λ-rpsL nupG tonA umuc:: pir116 -frt F′ (Lac+pro+ΔoriT::Tc)]为哈佛医学院 Stephen J Elledge教授所赠送[10],gfp模板质粒pHBM2002为本实验室朱德武博士构建[11]。
1.2 主要仪器和试剂
DNA分子量标准、SolutionⅠ连接试剂盒、LA Taq聚合酶和限制性内切酶NdeⅠ、XhoⅠ及StuⅠ购自TaKaRa公司;蛋白质分子量标准和限制性内切酶 BfuⅠ购于 Fermentas公司;质粒抽提试剂盒、琼脂糖凝胶回收试剂盒购自北京全式金生物技术有限公司,引物 (表 1)均在上海桑尼生物科技有限公司合成。所有的分子克隆操作都按照Sambrook等[12]提供的方法进行。
1.3 方法
1.3.1 定点诱变消除pET-23a(+)上的BfuⅠ位点
根据pET-23a(+)载体序列设计两对突变引物Mu23aF和Mu23aR、MublaF和MublaR,将载体上的两个 BfuⅠ酶切位点消除突变为 StuⅠ位点。PCR分别扩增BfuⅠ酶切位点两侧的载体片段,琼脂糖凝胶回收后共转化XL10-Gold感受态细胞,通过体内定点同源重组[11],获得重组中间载体 pET-23aM,送上海英骏生物公司测序。
1.3.2 构建定向T载体pETG
设计一对引物在5¢端各引入pET-23aM同源臂和一个BfuⅠ位点,下游引物紧邻BfuⅠ位点引入 13 bp的部分 LacO序列。用该引物从pHBM2002上扩增得到Prrn-gfp表达盒片段。载体PET-23aM经NdeⅠ和XhoⅠ双酶切后,琼脂糖凝胶回收大片段。将回收的载体片段与prrn-gfp表达盒片段共转化大肠杆菌 XL10-Gold感受态细胞,通过体内定点同源重组,获得定向T载体pETG,送上海英骏生物公司测序。
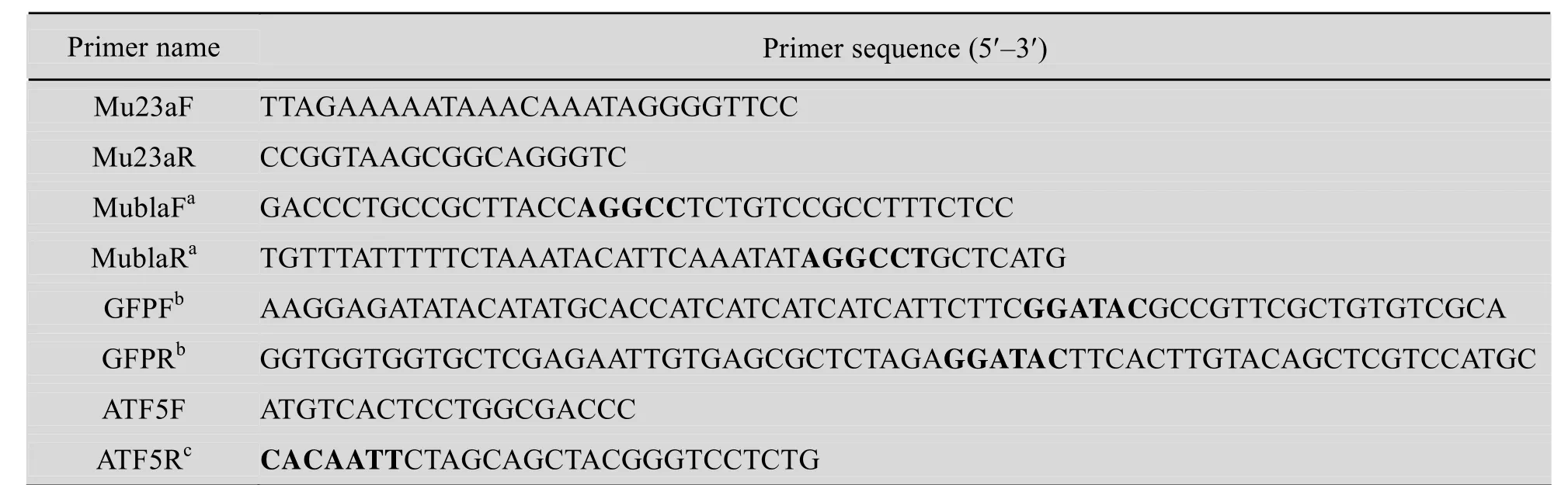
表1 引物序列Table 1 Primer sequences
1.3.3 BfuⅠ酶切制备T载体
BfuⅠ酶切载体pETG,琼脂糖凝胶回收完全酶切后的大片段,−20 ℃冻存。
1.3.4 PCR扩增目的基因和T载体克隆
根据NCBI数据库核苷酸序列查询,选择感兴趣的基因 (如ATF5),利用Genetool软件设计一对引物,在所有的下游引物5¢端额外补加7 bp部分 LacO 序列 (5¢-CACAATT-3¢,如表 1 所示)。利用该引物和 LA Taq DNA聚合酶扩增目的基因,并琼脂糖凝胶回收目的片段。用连接酶试剂盒连接~60 ng PCR产物和~20 ng线性化定向T载体,连接反应参见试剂盒说明。连接物转化大肠杆菌DH10β感受态细胞,涂布含有50 mg/mL氨苄西林钠和 25 mg/mL X-gal的 LB平板。在37 ℃培养箱中避光培养10 h后,挑取蓝色转化子抽提质粒。
1.3.5 蛋白质表达和SDS-PAGE检测
测序正确的克隆转化大肠杆菌 BL21(DE3)感受态细胞,蛋白质表达和SDS-PAGE检测方法参见分子克隆手册[12]。
1.3.6 目的蛋白质质谱鉴定
将目的蛋白质条带从SDS-PAGE胶上割下,按文献[13]进行胶内消化与质谱鉴定分析。
2 结果
2.1 定向T载体的构建
以质粒 pET-23a(+)为模板,引物 Mu23aF和Mu23aR、MublaF和MublaR分别扩增获得预期的~2.1 kb和~1.5 kb片段,琼脂糖凝胶回收后共转化大肠杆菌XL10-GOLD感受态细胞,获得重组中间载体 pET-23aM。由于实验设计是将两个BfuⅠ位点消除并诱变成StuⅠ位点,StuⅠ酶切鉴定 pET-23aM 可以切出约 1.5 kb小片段,而pET-23aM经BfuⅠ酶切无法切出小片段,该酶切鉴定结果表明pET-23a(+)上面的BfuⅠ酶切位点被成功诱变消除 (图2)。
以质粒 pHBM2002为模板,引物 GFPF和GFPR扩增获得预期的1 098 bp prrn-gfp表达盒片段,琼脂糖凝胶回收后通过同源重组插入pET-23aM 中 NdeⅠ和 XhoⅠ位点,获得定向 T载体pETG (图3)。pETG经BfuⅠ酶切得到约1 kb和3.6 kb的两个片段 (图4),该结果表明gfp表达盒已成功插入载体pET-23aM中,两个 BfuⅠ酶切位点均被正确引入。
2.2 制备线性化定向T载体
超纯质粒抽提试剂盒抽提pETG质粒,BfuⅠ完全酶切pETG后,琼脂糖凝胶回收约3.6 kb的T载体片段 (图4),制备线性化定向T载体完成,该T载体可以用于下游连接反应。

图2 酶切鉴定pET23aMFig. 2 Identification of pET23aM by restriction enzyme digestion. M: DNA marker; 1: pET23aM; 2:pET-23a(+); 3: pET23aM digested with Stu I; 4:pET23aM digested with Bfu I; 5: pET-23a(+)digested with Stu I; 6: pET-23a(+)digested with Bfu I.

图3 pETG载体图谱及部分关键序列Fig. 3 A plasmid map of pETG and key sequences. (A) gfp expression cassette flanked by 6×his tag-Bfu I sites and Bfu I sites-13/20 LacO sequence was inserted into Nde I-Xho I site of pET-23aM. (B) The key sequence of pETG was indicated. (C)Bfu I restriction site and a full length of LacO sequence were showed, N stands for any base.
2.3 连接转化和分析重组率
PCR扩增的基因片段连接线性化定向 T载体,转化大肠杆菌DH10β,从转化平板 (图1)上随机挑取 10个蓝色单菌落, 抽提质粒并进行酶切和PCR鉴定分析重组率。由于T载体末端设计有NdeⅠ和XhoⅠ酶切位点,可以利用该酶将插入片段ATF5 (849 bp)切除下来,质粒的酶切鉴定结果显示从随机挑取的 10个蓝色的单菌落抽提出来的质粒都切出目的基因大小的片段,与预期结果相符 (图 5A),同时利用基因特异性的上游引物和载体通用引物T7ter进行PCR扩增表明,10个质粒都含有目的基因插入,并且均为正向插入,扩增片段大小都和预期结果相符 (图5B),重组率为100%。
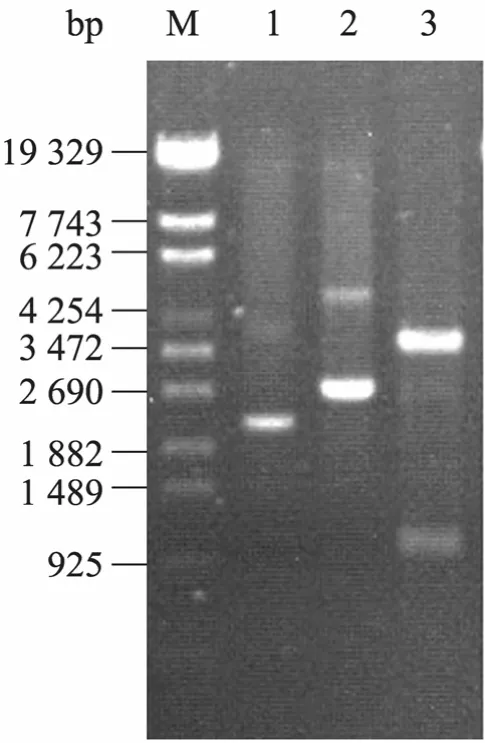
图4 定向T载体pETG琼脂糖凝胶电泳分析Fig. 4 Identification of directional T vector pETG by agarose gel electrophoresis. M: DNA marker; 1:pET23aM (3 666 bp); 2: pETG (4 649 bp); 3: pETG digested with Bfu I.
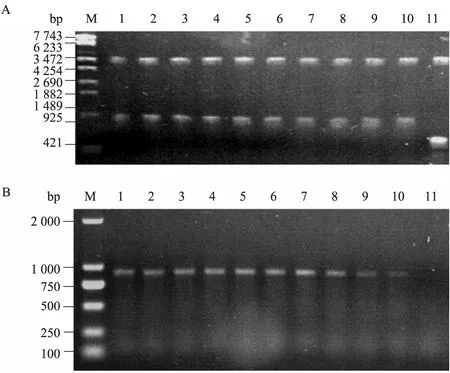
图5 重组子的酶切 (A) 和PCR (B)鉴定Fig. 5 Identification of recombinants by restriction enzyme digestion and PCR. (A)Identification of recombinants by Nde I-Xho I restriction digestion. M: DNA marker; 1−10: Nde I-Xho I digested plasmids isolated from 10 blue colonies,respectively; 11: PETG digested by Nde I-Xho I. (B)The plasmid DNA from 10 independent blue colonies was amplified by PCR, PCR products were separated on a 1.5% agarose gel and visualized after EB straining. M: DNA marker; 1−10: PCR products of 10 recombinants isolated from 10 blue colonies, respectively; 11: PCR product of vector PETG.
2.4 蛋白质表达和分析
本研究利用定向T载体成功地克隆了103个人类肝蛋白编码基因,从中随机挑选8个基因的克隆 (基因名称和克隆效率见表2),转化大肠杆菌BL21(DE3),IPTG诱导蛋白质表达后,SDSPAGE分析如图6所示,8个pETG克隆经诱导后在理论大小处均有蛋白条带变粗,表明有目标蛋白质表达 (图6),重组蛋白质条带割胶后经质谱鉴定证明为对应的目标蛋白质。
3 讨论
在当代分子生物学研究当中,无论研究的是已知基因还是未知基因,往往需要将扩增的目的基因克隆入适当的载体中,以便用于各种分析。T载体克隆 PCR产物虽然已经得到了广泛的应用,但是由于存在假阳性的问题,大量的资源和时间被浪费在后续重组子筛选鉴定中;对于某些基因的特定功能研究还需要进行蛋白质表达,由于基因插入T载体的方向是随机的,反向插入基因无法正常表达,理论上传统T载体克隆方法最高只有50%的几率获得定向插入的重组子,利用传统 T载体进行基因克隆和表达仍然需要耗费大量时间、人力和物力用来抽提质粒、鉴定基因的插入方向[7-8]。如何实现高通量基因克隆和表达,避免繁琐的筛选重组子工作、降低实验研究的工作强度一直是分子生物学研究者所希望的。
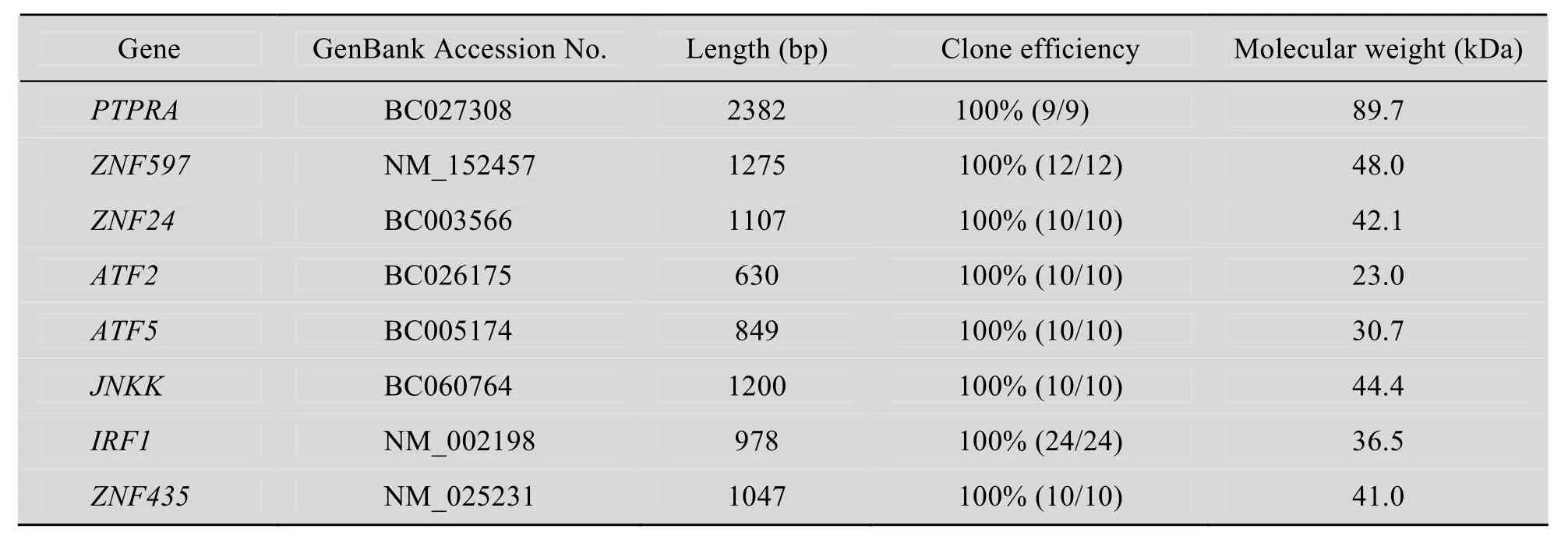
表2 已表达的8个基因的克隆效率和理论蛋白分子量Table 2 Clone efficiency and protein theoretical molecular weight of 8 gene

图6 SDS-PAGE分析重组蛋白Fig. 6 SDS-PAGE analysis of recombinant proteins. M: molecular mass standard protein. Control: BL21(DE3)/pETG.
目前利用传统的 T载体进行基因克隆存在两大问题,一是假阳性问题无法避免,需要大量抽提转化子进行鉴定;二是基因插入方向不能定向,仍然需要大量人工筛选转化子进行插入方向鉴定。传统方法利用GFP[14],KillerRed[8,15]或者LacZ[6]等基因的表型缺失来淘汰不完全酶切的载体背景,该方法在一定程度上降低了筛选重组子的工作量,但是仍然无法避免线性化T载体异常重组和反向插入基因造成的背景干扰;商品化的Topo®TA克隆试剂盒可以实现基因的定向克隆,重组效率大约在90%,该方法同样存在背景污染的问题,克隆仍然需要大量抽提质粒并鉴定重组子,并且载体只能一次性使用,需要购买昂贵的试剂盒。本研究利用自制的定向 T载体和PCR片段连接重构20 bp LacO序列来一步法表型筛选基因的插入和定向双重事件。当T载体和基因片段发生正确的连接反应时,T载体上的13 bp 部分LacO序列将和PCR片段上的7 bp部分LacO序列重构完整的20 bp LacO序列,重构的LacO序列将竞争性吸附宿主菌中的LacO结合蛋白 (LacI),对宿主菌的 LacZ基因表达去抑制 (图1),该单菌落在X-gal底物平板上将变蓝。该方法筛选重组子的方式比传统 T载体使用的LacZ alpha插入失活的遗传筛选方式更为严谨,T载体自环化产生的转化子、反向插入转化子、T载体背景和 LacZ自发移码转化子都不会被筛选到,而仅仅只有目的基因定向插入,且20 bp LacO序列成功重构的重组子可以被筛选到。由于该筛选方案是基于可见的表型获得筛选,不同于传统的表型缺失筛选,该筛选方法保证了克隆筛选的唯一正确性,如图1所示的蓝色转化子抽提质粒进行鉴定后显示定向插入重组效率为100%,所有被抽提的蓝色转化子质粒都含有定向插入的目的基因。
利用该定向T载体进行基因克隆非常简单、高效、可靠,不用购买商业的T载体试剂盒,还可以实现基因的定向插入;实验过程避免了繁琐的筛选鉴定重组子工作;并且该定向T载体还可以实现基因的无缝克隆,表达的蛋白质不会融合额外的非必要氨基酸序列。常规方法利用XcmⅠ、AhdⅠ、EcoR V制备T载体均会在克隆序列附近残留部分酶切位点碱基序列,无法实现无缝克隆,当进行蛋白质表达时,蛋白序列上会附加额外的氨基酸序列,该序列可能会对蛋白质的表达或者功能表征有一定负面影响。本研究利用限制性内切酶BfuⅠ酶切T载体实现了基因的无缝克隆,避免了非必要序列的融合。BfuⅠ属于IIS型限制性内切酶,其切割位点在识别位点之外,并且切割位点为识别位点下游第 5位任意的碱基(图 3C),通过 PCR扩增 prrn-gfp引入两个背向插入的BfuⅠ位点,其识别位点临近prrn-gfp,当制备线性化的T载体时,该限制性酶切位点被全部切除,不会残留任何酶切位点序列,从而实现了基因的无缝融合,当进行蛋白质表达时不会给蛋白质序列融合额外的氨基酸序列,保证了蛋白质序列的天然忠实性。
本实验成功构建了一种新颖的定向T载体,利用该T载体成功的定向克隆了103个人类肝蛋白编码基因。随机挑选了其中8个基因的克隆进行蛋白质表达显示8个基因对应的蛋白质均获得成功表达。利用该定向T载体进行基因的克隆和表达非常简单、高效,不仅省去了大量复杂的鉴定重组子工作,实现了基因的定向插入、基因的无缝融合表达,还可以节约大量宝贵的科研时间,降低克隆成本,同时该定向T载体还兼容传统的T载体基因克隆,有高通量大规模基因克隆和表达的应用前景。
[1]Eisenberg D, Marcotte EM, Xenarios I, et al.Protein function in the post-genomic era. Nature,2000, 405(6788): 823−826.
[2]Vukmirovic OG, Tilghman SM. Exploring genome space. Nature, 2000, 405(6788): 820−822.
[3]Cha J, Bishai W, Chandrasegaran S. New vectors for direct cloning of PCR products. Gene, 1993,136(1/2): 369−370.
[4]Jeung JU, Cho SK, Shim KS, et al. Construction of two pGEM-7Zf(+)phagemid T-tail vectors using Ahd I-restriction endonuclease sites for direct cloning of PCR products. Plasmid, 2002, 48(2):160−163.
[5]Holton TA, Graham MW. A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors. Nucleic Acids Res, 1991, 19(5):1156.
[6]Choi YJ, Wang TT, Lee BH. Positive selection vectors. Crit Rev Biotechnol, 2002, 22(3): 225−244.
[7]Ito Y, Suzuki M, Husimi Y. A T-extended vector using a green fluorescent protein as an indicator.Gene, 2000, 245(1): 59−63.
[8]Liu X, Liu X, Zhou Y, et al. T vector bearing KillerRed protein marker for red/white cloning screening. Anal Biochem, 2010, 405(2): 272−274.
[9]Cranenburgh RM, Lewis KS, Hanak JA. Effect of plasmid copy number and lac operator sequence on antibiotic-free plasmid selection by operator-repressor titration in Escherichia coli. J Mol Microbiol Biotechnol, 2004, 7(4): 197−203.
[10]Li MZ, Elledge SJ. MAGIC, an in vivo genetic method for the rapid construction of recombinant DNA molecules. Nat Genet, 2005, 37(3): 311−319.
[11]Zhu D, Zhong X, Tan R, et al. High-throughput cloning of human liver complete open reading frames using homologous recombination in Escherichia coli. Anal Biochem, 2010, 397(2):162−167.
[12]Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed. New York: Cold spring Harbor Laboratory Press, 2002.
[13]Chen X, Zhai C, Kang L, et al. High-level expression and characterization of a highly thermostable chitosanase from Aspergillus fumigatus in Pichia pastoris. Biotechnol Lett, 2012,34(4): 689−694.
[14]Chalfie M, Tu Y, Euskirchen G, et al. Green fluorescent protein as a marker for gene expression.Science, 1994, 263(5148): 802−805.
[15]Liu X, Shi R, Zou D, et al. Positive selection vector using the KillerRed gene. Anal Biochem, 2011 412(1): 120−122.
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
今日农业(2020年24期)2020-12-15 16:16:00
长春师范大学学报(2018年8期)2018-08-17 12:40:20
食品科学(2018年10期)2018-05-23 01:27:28
小学生导刊(2017年13期)2017-06-15 20:29:38
兽医导刊(2016年12期)2016-05-17 03:51:50
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
西南军医(2015年6期)2015-01-23 01:25:50
