
超正电荷绿荧光蛋白+36GFP作为核酸载体的细胞穿透性分析
2013-11-12 02:20:38李红玉房有荣于海涛俞盈阎辉
生物工程学报 2013年4期
李红玉,房有荣,于海涛,俞盈,阎辉
浙江省医学科学院药物研究所,浙江 杭州 310013
核酸转染哺乳动物细胞通常使用商业的阳离子脂质体试剂,然而,这些试剂的转染效率因细胞种类的不同差别很大。许多细胞株包括一些神经细胞、T细胞、成纤维细胞和上皮细胞等已证明抵抗一般的阳离子脂质体转染试剂[1-4]。另一些转染细胞的方法如电穿孔法[5]和病毒介导法[6-7]具有一定的细胞毒性或以未知方式扰乱细胞功能等缺点。近来出现的核酸转染新方法,包括lipidoids[8]、阳离子聚合物[9]、无机纳米粒子[10]、碳纳米管[11]、细胞穿透肽[12-13]、阳离子蛋白-抗体融合[14-15]、化学修饰的核酸[16]等,在针对特定种类细胞时,分别具有一定的优势。然而,通常这些方法准备过程较复杂,且在各细胞株中转染效率差别较大。
David R. Liu实验室通过研究绿色荧光蛋白(GFP)的分子表面结构,在不影响蛋白荧光特性的前提下,把非保守氨基酸替换成一些带正电荷或负电荷的氨基酸,得到一些“超电荷蛋白”[17]。这样的超电荷蛋白不仅保持原有的性能,而且获得了一些不同寻常的特性[18]。表面带有36个正电荷的绿色荧光蛋白 (+36GFP)由于静电排斥作用,使蛋白溶液具有更高的稳定性。+36GFP可以通过静电作用与DNA或RNA结合,形成单分散颗粒,用这种结合物转染细胞可以高效地将核酸携带进细胞[18]。诸多报道证明,很多带正电的多肽和蛋白都具有细胞穿透作用,例如衍生于艾滋病毒的tat细胞穿透肽[19-25]。+36GFP的转染效率是tat细胞穿透肽的100倍,对于一些耐受阳离子脂质体的细胞株也可以成功转染,而且无明显的细胞毒性[26]。因+36GFP保留了固有的绿色荧光特性,使整个实验操作可以肉眼观察,简化了操作过程。
改造得到的+36GFP所具有的这些特性,为寻找更理想的转染方法提供了新思路。这就为外源核酸进入细胞内发挥作用开辟了一条便捷之路,无论对科学研究还是对核酸药物的实际应用都具有非常重要的意义。本实验将初步研究+36GFP的这些特性,为以后进一步的研究打下基础。
1 材料与方法
1.1 材料
质粒pET+36GFP-HA2原核系统表达融合蛋白+36GFP-HA2,其中HA2为一段具有破囊泡作用的短肽,+36GFP蛋白N端具有His-tag标签,该质粒由哈佛大学的David R. Liu博士惠赠;质粒LeGO-1×T/BSD具有真核启动子,可在细胞中表达红荧光蛋白,由德国汉堡大学 Boris Fehse教授惠赠[27-28];感受态菌株 E.coli BL21(DE3)购自 Novagen公司;Ni-NTA Agarose购自QIAGEN公司;1 mL HiTrap Q FF离子层析柱、5 mL HiTrap Desalting脱盐柱购自GE公司;蛋白质分子量 Marker购自北京全式金公司和Fermentas公司;ECL显色液、FITC荧光标记试剂盒购自Pierce Biotechnology公司;牛血清白蛋白 (BSA)、LB培养基购自上海生工生物工程公司;DMEM、RPMI-1640、OPTI-MEM培养基、胎牛血清 (FBS)和 Lipofectamine 2000转染试剂均购自 Invitrogen公司;青霉素/链霉素双抗(10 000 IU/mL)购自天津灏洋生物;鼠抗His-tag一抗抗体购自天根公司、HRP标记的羊抗鼠IgG抗体均购于武汉博士德公司;BCA蛋白浓度测定试剂盒、考马斯亮蓝G-250染色液、脱色液购自碧云天公司。
1.2 主要仪器
二氧化碳细胞培养箱 (Sanyo,日本),蛋白电泳系统 (Bio-Rad,美国),荧光倒置显微镜(Leica,德国),流式细胞仪 (BD,美国),高速冷冻离心机 (Beckman,美国),激光共聚焦显微镜(ZEISS,德国)、凝胶成像仪 (Bio-Rad,美国)。
1.3 +36GFP蛋白的表达纯化
质粒pET+36GFP-HA2转化化学感受态表达菌株E. coli BL21(DE3),平板上挑取单克隆菌落,筛选高表达菌株。利用自动诱导培养基[29-30]表达目的蛋白,离心沉淀菌体,在非变性条件下裂解菌体。取上清按照 QIAGEN 公司 QIA expressionistTM蛋白纯化系统关于His-tag标签蛋白的纯化说明书操作[26]。将通过亲和层析得到的蛋白,先通过1 mL HiTrap Q FF离子层析柱进行离子交换,然后再通过5 mL HiTrap Desalting脱盐柱,用含10%甘油的PBS洗脱,过滤除菌。BCA法测定蛋白的浓度用于后续的实验。
1.4 +36GFP蛋白的细胞转导实验
B16细胞、293细胞、HepG2细胞用含有10% FBS和1%青霉素/链霉素双抗 (100 IU/mL)的 DMEM 培养基进行培养,A549细胞用含有10%FBS和1%青霉素/链霉素双抗 (100 IU/mL)的RPMI-1640培养基培养。将处于指数生长期的4种细胞分别传代于 24孔板,待细胞汇合度达90%时,用+36GFP蛋白进行转导。其中293细胞中+36GFP 的终浓度为 25、50、100、200 nmol/L;HepG2细胞中+36GFP的终浓度为25、50、100、200 nmol/L;A549细胞中+36GFP的终浓度为12.5、25、50、100 nmol/L;A549细胞中+36GFP的终浓度为12.5 nmol/L、25 nmol/L和50 nmol/L,同时设空白对照。37 ℃继续培养30 min后吸去培养液,用无钙镁溶液洗涤 1次,0.25%胰蛋白酶消化,用含20 U/mL肝素的PBS洗涤3次,重悬,流式细胞仪 FITC通道检测荧光细胞百分比。
接种A549细胞至2个NEST-35mm细胞培养皿中 (其玻底直径为 2 cm),待细胞融合度达到80%时,在其中1个平皿中加入+36GFP蛋白使其终浓度为100 nmol/L,另1个平皿不做处理,37 ℃孵育4 h后用含20 U/mL肝素的PBS洗涤3次,激光共聚焦观察+36GFP蛋白的转导效果。
1.5 +36GFP凝胶阻滞实验
用浓度为0.9 μg/μL的+36GFP蛋白和浓度为0.4 μg/μL 的质粒 LeGo-1×T/BSD 分别按 4∶1、9∶1、14∶1、19∶1和24∶1的体积比例混合。20 ℃振荡混匀30 min后,进行1%的琼脂糖凝胶电泳,凝胶成像仪中观察结果。
1.6 +36GFP蛋白结合质粒 LeGO-1×T/BSD转染HepG2细胞
接种HepG2细胞到4个NEST-35mm细胞培养皿中 (其玻底直径为 2 cm),次日细胞融合度达到80%时用于转染实验。其中1培养皿用脂质体 Lipofectamine2000转染质粒 LeGO-1×T/BSD(0.4 μg/μL):分别用 100 μL OPTI-MEM 培养基稀释 2 μg 质 粒 LeGO-1×T/BSD 和 5 μL Lipofectamine2000,室温孵育5 min后混合,继续孵育 20 min,弃去培养皿中的培养基用OPTI-MEM培养基洗涤细胞2次,将200 μL混合物加入平皿并再补加200 μL OPTI-MEM;另外3 个培养皿分别用 20 μL、50 μL、100 μL 的+36GFP蛋白结合2 μg质粒LeGO-1×T/BSD转染HepG2细胞:2 μg质粒LeGO-1×T/BSD分别与20 μL、50 μL、100 μL +36GFP 蛋白混合 20 ℃振荡混匀 30 min后加入平皿中,每个平皿用OPTI-MEM培养液补足至400 μL;最后1个培养皿中只加入2 μg质粒LeGO-1×T/BSD,同样用OPTI-MEM 培养液补足至 400 μL。所有平皿放入37 CO℃2孵箱里培养6 h,然后换液加入含有10% FBS无抗生素的 DMEM 培养基继续培养24 h。激光共聚焦观察细胞中质粒 LeGO-1×T/BSD的表达。
1.7 +36GFP蛋白携带质粒 LeGO-1×T/BSD转染293细胞
293细胞接种到12孔板中,待细胞融合度达到90%时用于转染实验。空白组为完全不做处理的细胞;实验对照组只加入4 μL质粒LeGO-1×T/BSD (0.4 μg/μL);脂质体转染组将 4 μL 质粒和4 μL Lipofectamine2000 分 别 加 入 100 μL OPTI-MEM培养基中,室温孵育5 min,混合继续孵育20 min,将200 μL混合物加入平板中;实验组共有 4组,+36GFP 蛋白 (0.9 μg/μL)和质粒分别按照25∶1、50∶1、100∶1和200∶1的体积比混合,20 ℃振荡混匀30 min后,加入培养孔中用 OPTI-MEM 培养基补足终体积为200 μL。平板放入37 CO℃2孵箱里培养6 h,然后换液加入含有 10% FBS无抗生素的 DMEM培养基。培养48 h后,0.25%胰蛋白酶消化,用含20 U/mL肝素的PBS洗涤3次,重悬,流式细胞仪FL2通道检测。
2 结果
2.1 +36GFP蛋白诱导表达的Western blotting验证
自动诱导培养基 30 ℃过夜培养含 pET+36GFP-HA2质粒的BL21(DE3)表达菌株,离心收集细菌沉淀,在紫外线下观察,与空白的BL21(DE3)菌株沉淀相比,具有很强的荧光 (未显示);Western blotting检测得到特异性的条带(图 1)。
2.2 +36GFP蛋白的表达纯化
大规模培养后,菌体沉淀裂解离心后,可直接观察到上清呈现绿色,提示目的蛋白在菌体内主要以可溶型而非包涵体形式存在。将此上清通过Ni-NTA柱子,可清楚地观察到蛋白与柱子结合,洗脱后洗脱液呈绿色。将亲和层析各步骤的样品进行SDS-PAGE蛋白凝胶电泳,考马斯亮蓝染色,脱色后发现+36GFP蛋白的纯度随着洗脱次数的增加纯度提高,最后一次洗脱几乎无其他杂带 (图 2)。

图1 +36GFP蛋白表达的Western blotting验证Fig. 1 Identification of the expressed +36GFP protein in E. coli by Western blotting. C: blank, bacterial with no plasmids; 1−4: bacterial colonies containing vector pET+36GFP–HA2, the primary antibody was mouse anti-His antibody and secondary antibody was goat anti-mouse IgG-HRP, and the positive band was visible at about 31 kDa; M: protein marker.
2.3 细胞转导实验
流式细胞仪分析显示,在293细胞、HepG2细胞、A549细胞和B16细胞中,低浓度的+36GFP蛋白即具有很高的转导效率,且随着浓度升高转导效率增加,呈现浓度依赖性,当浓度达到一定值时转导效率趋于稳定 (图3)。其穿透率的具体百分比在表1中列出。与对照组比较,激光共聚焦观察到+36GFP蛋白对 A549细胞的转导效果非常显著 (图4)。
2.4 凝胶阻滞实验
凝胶阻滞分析显示,+36GFP能够与带负电的质粒DNA结合,阻滞DNA在凝胶中迁移。图中明显观察到,随着+36GFP体积比例增加,蛋白对质粒的阻滞作用越来越大,呈现明显的浓度依赖性,最后DNA被完全地阻滞在了上样孔中 (图 5)。

图2 +36GFP蛋白通过His-tag亲和纯化各阶段样品的SDS-PAGE电泳Fig. 2 SDS-PAGE analysis of various stages samples from His-tag affinity chromatography of +36GFP protein. 1: the first washing; 2: the second washing; 3:the first elution; 4: the second elution; 5: the third elution; M: protein marker.
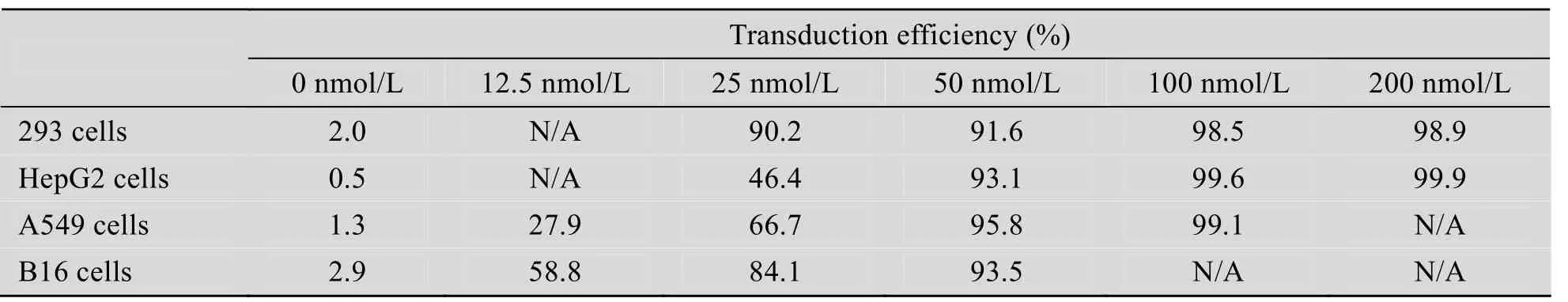
表1 +36GFP对各类细胞转导效率的流式细胞仪检测Table 1 Transduction efficiency of +36GFP protein to various cells tested by flow cytometry
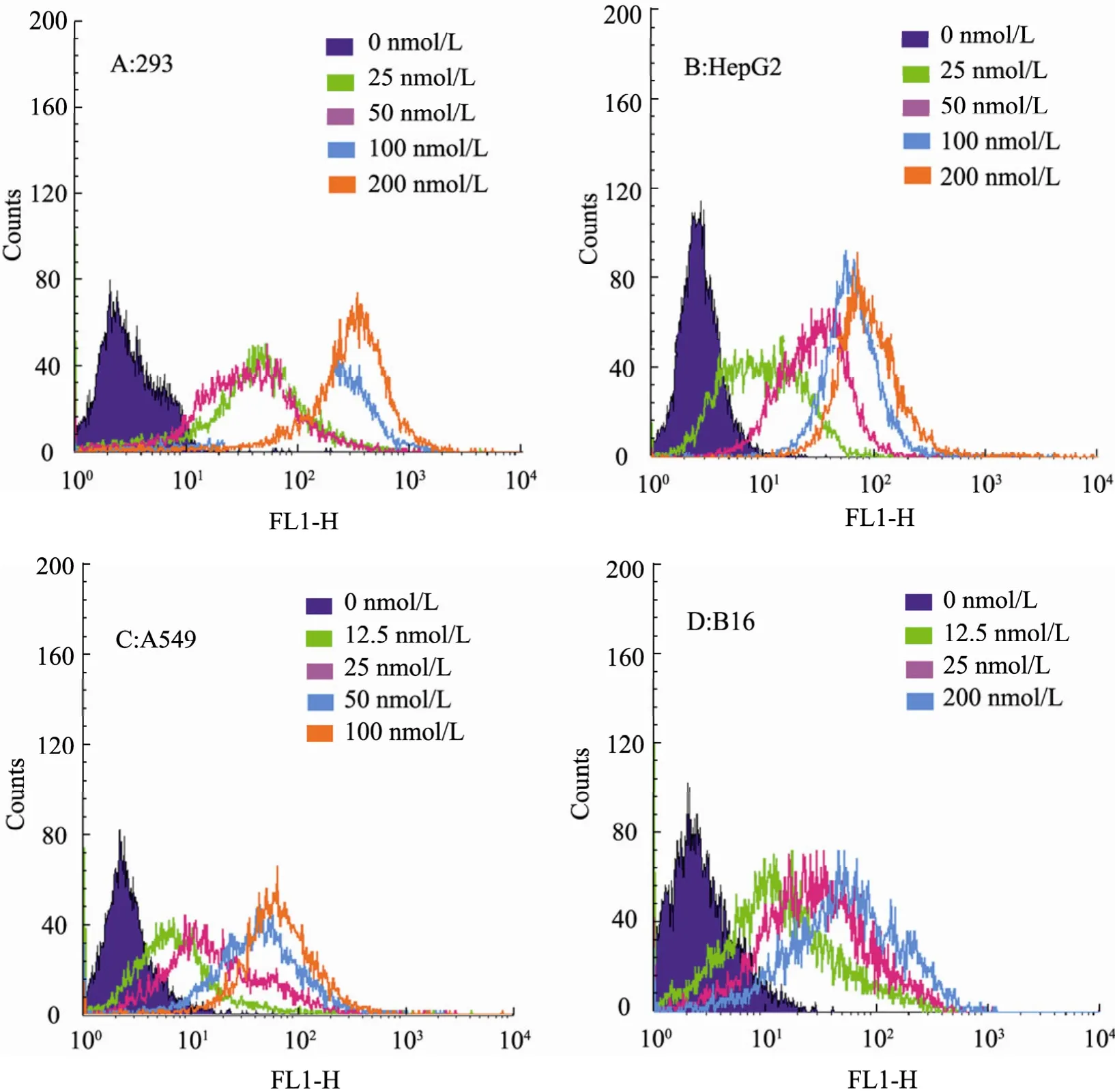
图3 +36GFP对各类细胞转导效率的流式细胞仪检测Fig. 3 Transduction efficiency of +36GFP protein to various cells assayed by flow cytometry. (A−D)The +36GFP protein was used to transduce a variety of mammalian cell lines including 293 cells, HepG2 cells, A549 cells and B16 cells at specified protein concentrations respectively. FACS analysis was performed on a BD Flow cytometry at 25 °C.

图4 +36GFP蛋白对A549细胞转导效率的激光共聚焦检测Fig. 4 Transduction efficiency of +36GFP protein to A549 cells observed under laser scanning confocal microscope.(A−C) The internalization of +36GFP in A549 cells after co-incubation for 4 h at 37 °C. (A) The image of UV. (B) The image of visible light. (C)The image is an overlay of two channels: white (visible light)and UV. (D−F)Control.
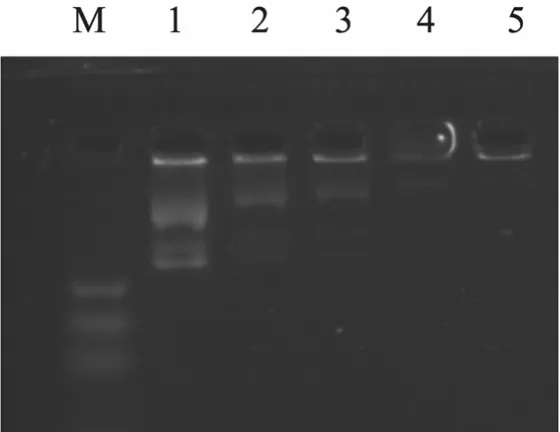
图5 +36GFP蛋白包裹质粒DNA的凝胶阻滞实验Fig. 5 Gel-shift assay of plasmid DNA packaged by+36GFP protein. M: DNA marker; 1−5: the volume ratio of +36GFP protein and plasmid DNA was 4:1, 9:1, 14:1,19:1 and 24:1, respectively.
2.5 HepG2细胞转染实验
激光共聚焦观察质粒 LeGO-1×T/BSD 的表达情况。该质粒经 Lipofectamine2000携带入细胞后,在 HepG2细胞中表达强度较高,在波长为540 nm的绿光下可观察到细胞中红荧光较强。20 μL、50 μL、100 μL 的+36GFP 蛋白也可携带质粒进入细胞,报告基因也可以表达,但是蛋白的表达量要低于脂质体转染组 (图6)。这可能是因为蛋白携带质粒进入细胞后要经历囊泡包裹、蛋白降解、质粒释放等步骤,这就使质粒表达红荧光的时间要晚于脂质体。至于+36GFP携带质粒转导细胞后蛋白何时表达量最高,是否呈现浓度依赖性,我们将进一步利用流式细胞仪技术证明。

图6 +36GFP蛋白对HepG2细胞转染效率的激光共聚焦检测Fig. 6 Transfection efficiency of +36GFP protein to HepG2 cells observed under laser scanning confocal microscope.(A) 5 μL Lipofectamine2000 transfection reagent transfected 2 μg plasmid LeGO-1×T/BSD (0.4 μg/μL) into HepG2 cells. (A1) The image is an overlay of two channels: white (visible light) and red (540 nm light). (A2) The image of 540 nm light. (A3) The image of visible light. (B−D) 20 μL, 50 μL and 100 μL +36GFP protein (0.9 μg/μL) with 2 μg plasmid LeGO-1×T/BSD (0.4 μg/μL). (B1, C1, D1) The overlay of two channels: white (visible light) and red (540 nm light). (B2, C2, D2)The image of 540 nm light. (B3, C3, D3)The image of visible light. (E)Control, only 2 μg plasmid,no Lipofectamine2000 and no +36GFP protein.
2.6 转染效率的流式细胞仪检测
流式细胞仪FL2-H通道检测293细胞中报告基因的表达情况。凝胶阻滞实验中,我们得出结论当蛋白与质粒的体积比大于24∶1,+36GFP蛋白可完全结合质粒LeGO-1×T/BSD阻滞其在凝胶中的迁移。因而此实验中我们设计了4组实验,蛋白与质粒的体积比依次是25∶1、50∶1、100∶1和200∶1,流式检测结果显示红荧光蛋白的表达量随着体积比的增大而增高 (图 7A)。相比较于HepG2细胞转染实验,该实验中转染时间从 24 h延长至48 h,梯度拉大,流式细胞仪统一计数10 000个细胞中有红荧光表达的细胞所占的百分比,随着体积比增大百分比依次为 55.22%、73.58%、85.46%和89.24% (图7B),呈现剂量效应关系。
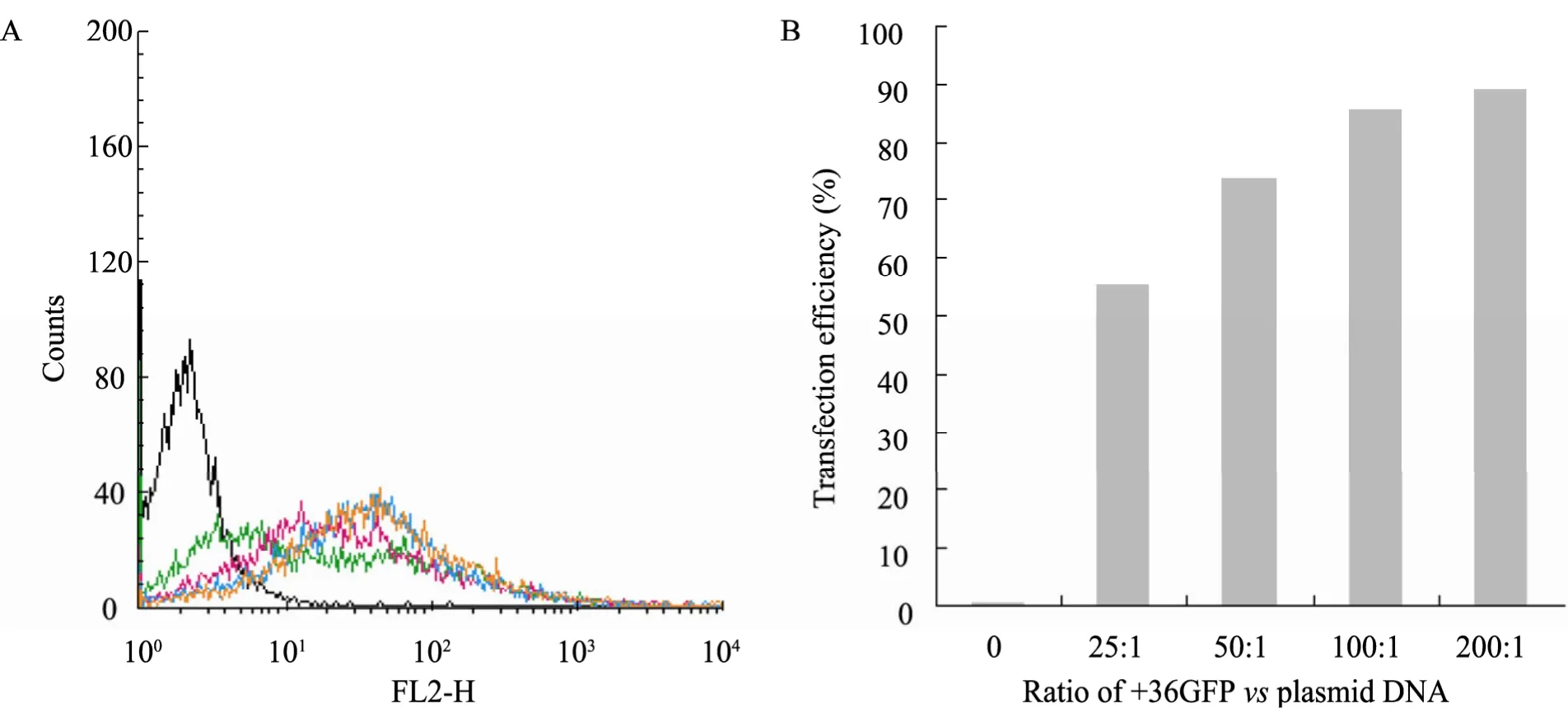
图7 +36GFP蛋白对293细胞转染效率的流式细胞检测Fig. 7 Transfection efficiency of +36GFP protein to 293 cells tested by flow cytometry. (A)Flow cytometry analysis showing amounts of expressed red fluorescent protein in 293 cells treated with various ratio of +36GFP vs plasmid DNA and washed three times with PBS containing heparin to remove cell surface-bound GFP. (B)Histogram of the expressed red fluorescent protein.
3 讨论
1990年 Wolff等发现,将表达报道基因的质粒直接注入骨骼肌后能检测到报道基因的表达[31],但直接注射裸露的DNA或质粒DNA进入细胞,DNA的表达效率很低;与直接注射裸DNA相比,采用电穿孔法可使DNA的表达效率高1 000倍[32],但对细胞损害较大;微粒轰击法,即“基因枪”,对目的DNA没有要求,可大可小,也可以是RNA、蛋白质、化学药物等多种物质,且无明显的致细胞变性反应,但其对实验条件的要求相对较高[33]。目前实验室应用最广泛的是阳离子脂质体转染。外源DNA与脂质体结合,形成载体-DNA复合物,利用这种复合物将外源DNA导入靶细胞,最终使外源基因在靶细胞中获得表达,但阳离子脂质体对细胞具有一定的毒性且转染效率因细胞不同而有所差异。
本研究成功表达并纯化了+36GFP蛋白,并证实+36GFP确有很强的细胞穿透性,而且细胞种类差异性很小。+36GFP穿透细胞膜的机理,国外许多科研人员已经进行了相应的研究[34]。+36GFP蛋白自身带有36个静电荷,相互存在很强的静电排斥作用而不容易聚沉,蛋白溶液更为稳定,不仅方便了整个蛋白表达纯化的操作过程,而且有利于蛋白保存、蛋白功能研究及进一步的开发应用。根据脂质体转染原理,带正电的脂质体颗粒可以包裹带负电的质粒分子,然后将其携带入细胞。+36GFP表面带有36个正电荷,同样可以与带负电的核酸分子结合,充当一种转染试剂。本研究中,我们首先进行了凝胶阻滞实验,验证了当蛋白和质粒DNA相互间达到一定的比例时,蛋白会完全将DNA包裹住,阻滞其在凝胶中的迁移。HepG2转染实验证实脂质体和+36GFP蛋白都可携带质粒进入细胞,而且质粒上的红荧光蛋白可以成功表达。在实验的过程中,我们发现脂质体转染会对细胞造成一定的细胞毒作用,而+36GFP蛋白对细胞的生长几乎没有影响。激光共聚焦图片中可观察到,+36GFP转染的红荧光蛋白的表达量要低于脂质体。我们分析出现这种现象的原因可能是+36GFP蛋白携带质粒进入细胞后,+36GFP-质粒复合物要经历一个囊泡包裹和蛋白降解的过程,之后质粒被释放红荧光蛋白得以表达,这就使得+36GFP转染需要的时间大于脂质体转染。为此我们又设计了293细胞转染实验,依次增高蛋白相对质粒的比例,然后把转染时间从24 h延长至48 h,得到了预期的结果。随着比例的增加,红荧光蛋白的表达量呈现剂量效应关系。
理想的转染方法应该具有价廉易得、性质稳定、转染率高、没有细胞种类差异、没有细胞毒性等特点,而本文中+36GFP蛋白似乎为我们找到一种理想的转染方法带来了一些新启发。除了寻找理想转染方法,+36GFP蛋白的许多特性也使我们看到了它巨大的应用前景。其实David R.Liu博士这种通过替换蛋白非保守氨基酸,重构蛋白表面电荷的方法,也可以应用于其他蛋白,可能为研究其他蛋白并赋予其细胞穿透性及抗凝聚等新特征提供一种非常新颖又有效的策略。
[1]Carlotti F, Bazuine M, Kekarainen T, et al.Lentiviral vectors efficiently transduce quiescent mature 3T3-L1 adipocytes. Mol Ther, 2004, 9(2):209−217.
[2]Ma H, Zhu J, Maronski M, et al. Non-classical nuclear localization signal peptides for high efficiency lipofection of primary neurons and neuronal cell lines. Neuroscience, 2002, 112(1):1−5.
[3]McManus MT, Haines BB, Dillon CP, et al. Small interfering RNA-mediated gene silencing in T lymphocytes. J Immunol, 2002, 169(10):5754−5760.
[4]Strait KA, Stricklett PK, Kohan JL, et al. Calcium regulation of endothelin-1 synthesis in rat inner medullary collecting duct. Am J Physiol Renal Physiol, 2007, 293(2): F601−606.
[5]Jantsch J, Turza N, Volke M, et al. Small interfering RNA (siRNA)delivery into murine bone marrow-derived dendritic cells by electroporation. J Immunol Methods, 2008, 337(1):71−77.
[6]Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell, 2002, 2(3): 243−247.
[7]Stewart SA, Dykxhoorn DM, Palliser D, et al.Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA, 2003, 9(4): 493−501.
[8]Akinc A, Zumbuehl A, Goldberg M, et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol,2008, 26(5): 561−569.
[9]Segura T, Hubbell JA. Synthesis and in vitro characterization of an ABC triblock copolymer for siRNA delivery. Bioconjug Chem, 2007, 18(3):736−745.
[10]Sokolova V, Epple M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew Chem Int Ed Engl, 2008, 47(8): 1382−1395.
[11]Liu Z, Winters M, Holodniy M, et al. siRNA delivery into human T cells and primary cells with carbon-nanotube transporters. Angew Chem Int Ed Engl, 2007, 46(12): 2023−2027.
[12]Deshayes S, Morris MC, Divita G, et al.Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci, 2005,62(16): 1839−1849.
[13]Meade BR, Dowdy SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides.Adv Drug Deliv Rev, 2008, 60(4/5): 530−536.
[14]Peer D, Zhu P, Carman CV, et al. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc Natl Acad Sci USA, 2007, 104(10): 4095−4100.
[15]Song E, Zhu P, Lee SK, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol, 2005, 23(6):709−717.
[16]Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with 'antagomirs'. Nature,2005, 438(7068): 685−689.
[17]Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc, 2007, 129(33): 10110−10112.
[18]McNaughton BR, Cronican JJ, Thompson DB, et al. Mammalian cell penetration, siRNA transfection, and DNA transfection by supercharged proteins. Proc Natl Acad Sci USA,2009, 106(15): 6111−6116.
[19]Daniels DS, Schepartz A. Intrinsically cell-permeable miniature proteins based on a minimal cationic PPII motif. J Am Chem Soc,2007, 129(47): 14578−14579.
[20]Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell,1988, 55(6): 1189−1193.
[21]Fuchs SM, Raines RT. Arginine grafting to endow cell permeability. ACS Chem Biol, 2007, 2(3):167−170.
[22]Fuchs SM, Rutkoski TJ, Kung VM, et al.Increasing the potency of a cytotoxin with an arginine graft. Protein Eng Des Sel, 2007, 20(10):505−509.
[23]Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein.Cell, 1988, 55(6): 1179−1188.
[24]Smith BA, Daniels DS, Coplin AE, et al. Minimally cationic cell-permeable miniature proteins via alpha-helical arginine display. J Am Chem Soc,2008, 130(10): 2948−2949.
[25]Thoren PE, Persson D, Karlsson M, et al. The antennapedia peptide penetratin translocates across lipid bilayers-the first direct observation. FEBS Lett, 2000, 482(3): 265−268.
[26]Cronican JJ, Thompson DB, Beier KT, et al. Potent delivery of functional proteins into Mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem Biol, 2010, 5(8): 747−752.
[27]Weber K, Bartsch U, Stocking C, et al. A multicolor panel of novel lentiviral "gene ontology"(LeGO)vectors for functional gene analysis. Mol Ther, 2008, 16(4): 698−706.
[28]Weber K, Mock U, Petrowitz B, et al. Lentiviral gene ontology (LeGO)vectors equipped with novel drug-selectable fluorescent proteins: new building blocks for cell marking and multi-gene analysis.Gene Ther, 2010, 17(4): 511−520.
[29]Lu Z, Chen W, Liu R, et al. A novel method for high-level production of psychrophilic TAB5 alkaline phosphatase. Protein Expr Purif, 2010,74(2): 217−222.
[30]Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif,2005, 41(1): 207−234.
[31]Wolff JA, Malone RW, Williams P, et al. Direct gene transfer into mouse muscle in vivo. Science,1990, 247(4949): 1465−1468.
[32]Blair-Parks K, Weston BC, Dean DA. High-level gene transfer to the cornea using electroporation. J Gene Med, 2002, 4(1): 92−100.
[33]Yang NS, Sun WH, McCabe D. Developing particle-mediated gene-transfer technology for research into gene therapy of cancer. Mol Med Today, 1996, 2(11): 476−481.
[34]Thompson DB, Villaseñor R, Dorr BM, et al.Cellular uptake mechanisms and endosomal trafficking of supercharged proteins. Chem Biol,2012, 19(7): 831−843
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19 13:59:46
陶瓷学报(2021年1期)2021-04-13 01:33:02
军事文摘(2020年20期)2020-11-16 00:31:56
中学生数理化·八年级物理人教版(2020年12期)2020-01-01 15:22:24
中学生数理化·八年级物理人教版(2018年12期)2019-01-31 02:38:18
食品科学(2018年10期)2018-05-23 01:27:28
中成药(2018年2期)2018-05-09 07:20:08
中成药(2017年3期)2017-05-17 06:08:52
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
