
纤维素酶家族及其催化结构域分子改造的新进展
2013-09-03 19:18张小梅李单单王禄山赵越陈冠军
生物工程学报 2013年4期
张小梅,李单单,王禄山,赵越,陈冠军
山东大学生命科学学院 微生物技术国家重点实验室,山东 济南 250100
纤维素酶家族及其催化结构域分子改造的新进展
张小梅,李单单,王禄山,赵越,陈冠军
山东大学生命科学学院 微生物技术国家重点实验室,山东 济南 250100
张小梅, 李单单, 王禄山, 等. 纤维素酶家族及其催化结构域分子改造的新进展. 生物工程学报, 2013, 29(4): 422−433.
Zhang XM, Li DD, Wang LS, et al. Molecular engineering of cellulase catalytic domain based on glycoside hydrolase family.Chin J Biotech, 2013, 29(4): 422−433.
纤维素酶的分子改造是其催化性能改进及催化效率提升的重要手段。近年来,组学技术与结构测定技术的迅速发展,人们已建立了包括糖苷水解酶 (Glycoside hydrolase,GH) 在内的碳水化合物活性酶组分数据库。通过对同一蛋白家族进行序列比对、分子进化分析与祖先基因重构,以结构模建分析为指导的纤维素酶分子改造,可以明显缩小序列或结构的搜索空间,加快酶分子改造的速度,增大理性设计成功的概率;同时针对催化中心活性架构的分析可以进一步阐明纤维素酶的催化机理与酶分子持续性降解机制。文中主要对纤维素酶家族及其催化结构域的分子改造取得的最新进展作了综述。在后基因组时代基于蛋白质家族中的海量数据分析,以其保守结构信息为指导的理性设计,将会成为纤维素酶分子改造的重要方向,从而推动生物质转化工艺的快速发展。
纤维素酶,糖苷水解酶家族,结构生物信息学,分子改造,理性设计
纤维素是构成植物细胞壁的重要组分,是目前地球上含量最丰富的可再生性资源[1]。纤维素酶属于糖苷水解酶类,是专门催化水解纤维素链中 β-1,4-糖苷键的一类酶的总称。通过纤维素酶对纤维素类生物质进行生物转化,获得相应生物基产品及生物燃料,对解决能源危机及促进社会可持续发展具有重要的战略意义。但由于现有纤维素酶降解效率偏低、生产成本较高等因素限制了其在工业上的广泛应用[2]。通过分子生物学与蛋白质工程手段进行纤维素酶的分子改造,以改变其催化活性或其他反应特性(如pH适应范围、热稳定性及持续催化能力等),不仅可以阐明天然纤维素酶的降解机制及其结构与功能的进化关系,同时也为进一步对其进行理性设计与改造,以获得能够满足生物质高效转化的纤维素酶组分提供可能[3]。
1 纤维素酶家族及其功能演化
1.1 纤维素酶基因序列的多样性与拓扑结构的保守性
纤维素酶主要是由各类微生物产生的,其中丝状真菌所产的典型游离纤维素酶分子具有多结构域的构架,包括碳水化合物结合模块(Cellulose-binding module,CBM),其通过柔性的连接肽 (Linker) 连接至催化结构域 (Catalytic domain,CD) 上,另外部分酶分子还含有其他结构域。构成厌氧细菌纤维小体的纤维素酶分子一般由一个对接模块 (Dockerin) 通过连接肽与一个催化结构域 (CD) 连接,其中对接模块可与支架蛋白(Scaffoldin,又称作脚手架蛋白)的粘连模块 (Cohesin) 结合,而支架蛋白一般还会存在一个CBM[4]。CBM在结合及降解天然纤维素的过程中具有重要作用[5];而CD则主要是催化糖苷键的断裂。
根据蛋白质结构域中氨基酸序列的相似性,将不同物种来源的碳水化合物活性酶类(Carbohydrate-Active enZYmes,CAZy)分成不同的蛋白质家族 (http://www.cazy.org/)。其中糖苷水解酶现已有131个家族,纤维素酶类分布在至少17个GH家族中,是糖苷水解酶数据库中家族数目最多的一类水解酶类[6]。不同家族的纤维素酶具有不同的进化起源与不同的演化历史,有的 GH家族亲缘关系较远 (Deeply rooted evolutionarily),如 GH9家族的纤维素酶,产生该类酶的生物种类广泛,有细菌 (包括好氧菌和厌氧菌)、真菌、植物与动物 (原生动物与白蚁),另外GH5与GH12家族的成员也广泛分布于古菌、细菌与真菌三类生物中;有的家族亲缘关系较近 (Lightly rooted evolutionarily),如GH7家族只含有真菌的水解酶类,GH8家族只含有细菌产生的水解酶类等等。有的家族尽管其亲缘关系较近,但功能的演化却非常迅速,如 GH7家族除含有内切纤维素酶外,还含有高效降解结晶纤维素的外切纤维素酶类[7]。另外,从微生物基因组角度分析,降解纤维素的微生物不同,其基因组中含有的纤维素酶家族也是不同的。从现已进行全基因组测序的1 500余种微生物中发现,其中40%以上的微生物其基因组中至少含有一种纤维素酶基因;而在高效降解纤维素的微生物基因组中,则一般会含有几十条编码纤维素酶的基因,且这些基因分属不同的GH家族。如隶属于丝状真菌的瑞氏木霉Trichoderma Reesei(红褐肉座菌Hypocrea jecorina的无性型),其分泌的纤维素酶主要分布于 GH5,GH6,GH7,GH12,GH45与GH61家族;放线菌中的褐色高温单孢菌Thermobifida fusca主要有来自GH5,GH6,GH9与GH48家族的相关纤维素酶基因;而好氧细菌中哈氏噬纤维菌Cytophaga hutchinsonii主要产生GH5与GH9家族的相关纤维素酶[8];厌氧细菌中的热纤梭菌Clostridium thermocellum主要产生GH5,GH8,GH9与GH48家族的相关蛋白[9]。
目前对 CAZy家族的分类是基于蛋白质氨基酸序列相似性进行的,分类的基础依据是序列相似度高于30%的即被归为同一GH家族。由于蛋白质的氨基酸序列与其折叠结构具有一定关系,故若蛋白质的氨基酸序列相似,其就有可能具有相同或相似的拓扑折叠结构,尤其是其活性部位的拓扑结构。这种以序列相似性为依据的 GH家族分类,为研究同一家族内某一未知蛋白的结构及催化机制提供了重要的信息。另外,基于拓扑结构的相似性,又将不同纤维素酶家族归于相同的“族系 (Clan)”。根据水解过程中产物糖分子异头碳羟基立体化学结构的变化,将纤维素酶的催化断键机制分为保留型或翻转型两种类型[10]。同一家族具有相同的催化断键机制;同一族系,甚至不同族系都可能会具有相同的断键机制,表 1列出了部分主要纤维素酶家族的蛋白结构折叠类型、催化机制及其他主要信息。
1.2 纤维素酶家族结构与功能的演化
根据纤维素酶降解底物时不同的作用方式可将其分成 3类:1) 内切纤维素酶又称之为内切葡聚糖酶 (Endoglucanase,EG;EC 3.2.1.4);2) 外切纤维素酶又称之为纤维二糖水解酶(Cellobiohydrolase,CBH;EC 3.2.1.91,EC 3.2.1.176);3) β-葡萄糖苷酶 (β-glucosidase,BGL;EC 3.2.1.21)。表2列出了GH家族中不同功能纤维素酶分子的分布情况。不同家族具有不同的蛋白质结构折叠类型,而不同蛋白结构进化出相同功能活性的酶分子,这说明蛋白功能进化的收敛性。从其家族分布来看,内切纤维素酶分布最广,外切纤维素酶分布于内切纤维素酶家族之中,β-葡萄糖苷酶仅在GH5和GH9家族与内切/外切纤维素酶相关家族有交叠。同一蛋白质家族具有相同的蛋白质拓扑折叠结构,而相同的折叠结构又有着明显不同功能的酶组分,说明同一家族的蛋白功能在进化过程中发生了分歧演化。因此通过研究蛋白质家族内酶分子功能演化的结构基础,将有助于对蛋白质进行有目的的理性设计。
大量纤维素酶结构研究表明,内切纤维素酶CD的活性部位是一个开放的裂隙 (图1A),可结合于纤维素链上,随机切断非结晶区的糖链,产生系列寡糖与新的还原末端。外切纤维素酶 CD的活性部位是一条孔道,纤维素的一条单链通过穿过该孔道而与CD结合,沿着纤维素链向结晶区单方向持续性催化 (Processive catalysis),产物一般为纤维二糖,因此又称为纤维二糖水解酶。根据结合分子链末端的不同,又将外切纤维素酶分成两类:一类作用于纤维素分子的非还原端(EC 3.2.1.91),这类酶的典型代表是GH6家族的纤维素酶分子 (图 1B);另一类作用于纤维素链的还原端 (EC 3.2.1.176),该类型的代表是由好氧真菌产生的GH7家族的纤维素酶分子 (图1C)及由厌氧细菌产生的GH48家族酶分子 (图1D)。已报道具有持续性降解作用的内切纤维素酶多属GH9家族 (图1E),其活性部位与一般内切纤维素酶相似,仍然是一开放裂隙,该类纤维素酶的催化结构域与 CBM 的连接肽很短,其 CBM为第3家族,研究表明,该种结构对于这类酶分子的持续性降解是必需的[11]。
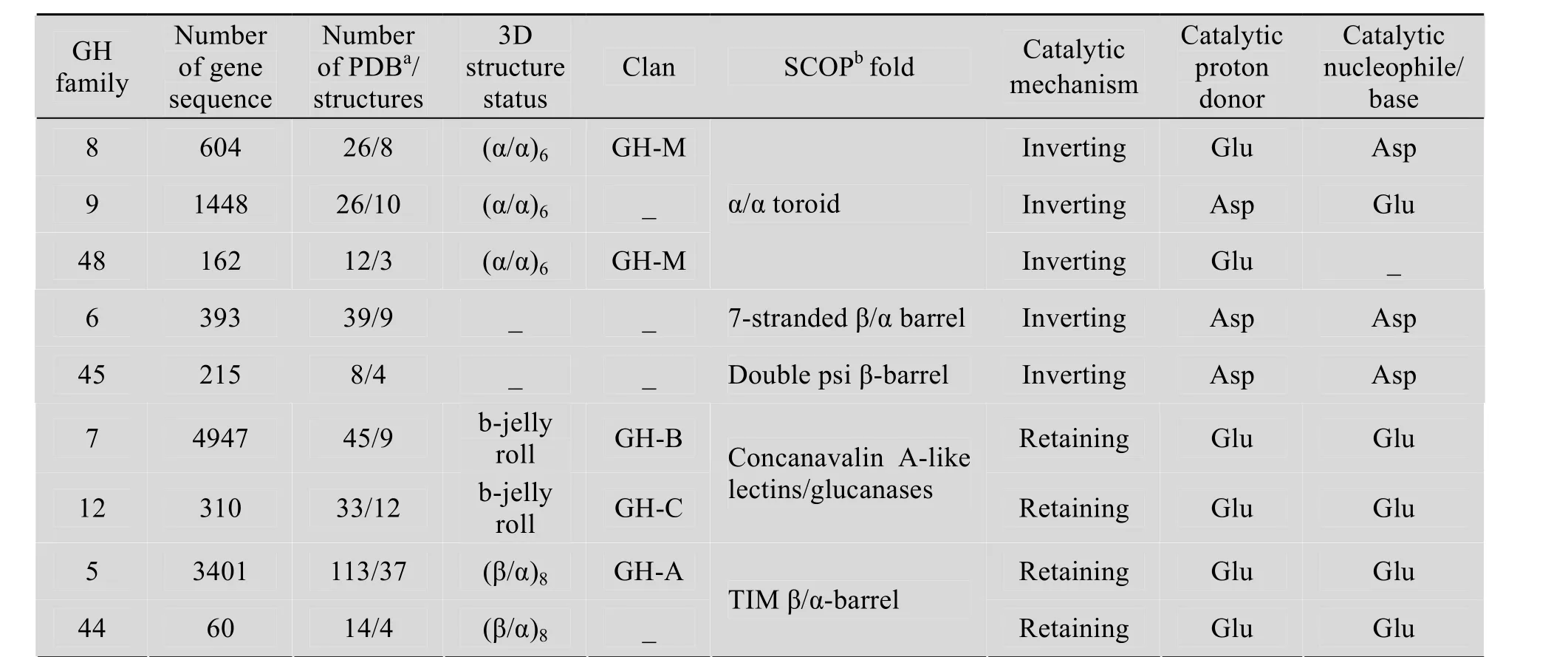
表1 主要纤维素酶家族*的基因序列、拓扑结构、催化机制及其成员信息统计表**Table 1 Statistics on the gene sequence, topological structure, catalytic mechanism and other information of the main glycoside hydrolase families*

表2 纤维素酶相关GH家族中不同功能糖苷水解酶的分布*Table 2 Distributions of glycoside hydrolases with different function in each cellulase relating GH family*

图1 不同功能纤维素酶三维空间结构示意图Fig. 1 3D structures of different cellulases. (A) T. reesei Cel7B (PDB ID 1EG1). (B) T. reesei Cel6A (PDB ID 1QK2).(C) T. reesei Cel7A (PDB ID 1CEL). (D) C. thermocellum Cel48A (PDB ID 1L1Y). (E) T. fusca Cel9A (PDB ID 1JS4).Catalytic residues are shown in sticks (blue).
同一家族纤维素酶的分子结构相对保守,但在长期的进化过程中却演化出适应不同环境的序列组成及功能特征。对纤维素酶家族序列与结构的初步分析发现,插入/缺失可以导致同一家族纤维素酶分子功能发生快速演化,并且该规律可推广至多数蛋白质家族;另外,SCOP数据库(http://scop.mrc-lmb.cam.ac.uk/scop/) 中多数同源结构家族中单氨基酸替换和插入/缺失对蛋白质结构的进化具有共同作用[12];蛋白结构发生插入/缺失后其侧翼区域的二级及三级结构变化明显,同时可以降低侧翼区域的选择压力,这是功能快速演化的结构基础[13-14]。所以针对家族内酶分子的进化分析,有助于认识其序列空间多样性与结构空间有限性之间的关系,可以快速找出功能演化的决定因素,为后期进行酶分子改造提供全新思路[15]。
1.3 高效降解结晶纤维素的持续性外切酶类
木质纤维素生物质在长期的进化过程中形成了复杂的超分子结构,限制了微生物及相应酶分子的有效降解,现在统称为“生物质抗降解屏障”[2],其中绿色植物结晶纤维素是主要的降解限制因素之一。在纤维素合成时,由纤维素合成酶系的末端复合体 (Terminal complex,TC) 同时完成36根葡萄糖链的合成,形成3 nm×5.5 nm左右的结晶状微纤丝也称之为基元纤丝[2]。纤维素在酶解过程中,相应的酶分子要从微纤丝上剥离单根糖链,这被认为是限制纤维素水解的关键步骤。内切纤维素酶随机定位在微纤丝上的非结晶区,产生大量还原端,而外切纤维素酶结合于单根纤维素链的还原端或非还原端,持续性地降解结晶区的纤维素分子链。由于结晶纤维素在木质纤维素中含量较高,所以持续降解的外切纤维素酶是降解结晶纤维素的主要酶类,研究表明其活性的丧失被认为是纤维素酶解过程中速率降低的主要原因[16]。因此纤维素酶理性设计的重要目标之一就是设计新型的外切纤维素酶类,尽量提高其降解结晶纤维素的效率。
外切纤维素酶催化结构域的孔道结构使其在释放纤维二糖产物的同时,仍能够紧紧地结合于纤维素链上,以便完成单方向持续性地催化水解。已有实验证实,纤维二糖水解酶的可持续性催化动力主要由糖苷键水解所释放的能量驱动[17]。该过程有两步基元反应,计算获得释放的能量为−26.37 kcal/mol[18]。酶分子与底物的相互作用可从晶体表面上剥离出单根纤维素链,纤维二糖水解酶的这种催化方式类似于 DNA解旋酶,后者则主要是利用ATP的水解释放的能量使其沿DNA链持续前进并打破双螺旋间的氢键[19]。像这样可以转化化学能或热能做功的生物大分子,通常被称为“分子机器”[20]。研究“分子机器”的序列、结构、分子动态学及其功能,在空间与时间上描述酶分子催化过程的所有原子特征,可以解释与预测酶分子持续性催化的机理与功能,有利于对外切纤维素酶类进行理性设计。
2 纤维素酶的分子改造
自从 1983年 Ulmer提出“蛋白质工程(Protein Engineering)”概念以来,纤维素酶的分子改造先后经历了以下 3个阶段:1) 以定点突变为基础的“定点理性设计”;2) 以易错PCR或DNA改组技术主导的“定向进化”;3) 目前以数据驱动设计或结构生物信息学指导的“结构理性设计”[21]。
2.1 以定点突变为代表的定点理性设计
20世纪80年代,随着定点突变技术的快速发展,纤维素酶理性改造策略应运而生。其基本概念是首先选择一合适的纤维素酶分子,选定特定的氨基酸进行突变,对相关蛋白进行性质研究。该方法对阐明酶分子活性部位的关键氨基酸与催化断键机制,以及对其催化或与底物结合有关的关键位点的鉴定是非常有帮助的。通过定点突变技术已成功分析了不同芽胞杆菌属来源的纤维素酶具有不同最适催化 pH的原因;Zhang等对褐色高温单孢菌T. fusca来源的Cel6A活性位点周围的关键氨基酸进行了定点突变,提高了其催化降解羧甲基纤维素 (Carboxymethyl cellulose,CMC) 的活力,增强了对该底物的专一性;在以天然纤维素为底物的研究中,2005年Barker等对一嗜酸耐热解纤维菌Acidothermus cellulolyticus的内切纤维素酶Cel5A第245位进行定点突变 (Y245G),降低了末端产物的抑制作用,使其对微晶纤维素的降解活力提高了20%[22]。
另外,定点改造技术还证明了外切纤维素酶活性部位特有的凸环 (Loop) 结构在可持续降解过程中发挥了重要作用。若定向删除粪肥纤维单胞菌Cellulomonas fimi来源的Cel6B (之前称为Cellobiohydrolase A) C末端的一段凸环结构,其对可溶性底物 (如 CMC) 的降解能力则有所提高。对褐色高温单孢菌T. fusca中具有可持续降解能力的Cel6B的6个凸环结构分别进行定点突变,发现所有突变体对滤纸与细菌微晶纤维素(Bacterial microcrystalline cellulose,BMCC) 的降解能力都有所降低,同时其可持续性也有所减弱,据此推测这些凸环结构在Cel6B的可持续性降解中起着重要作用。另外,Von对瑞氏木霉Cel7A的凸环结构中 3个位点 (D241,Y247和D249) 进行定点突变,通过消除酶分子与底物间的氢键及引入二硫键的方法,提高了酶分子对无定型纤维素与结晶纤维素的降解活力[22]。
尽管定点突变技术已经成功地应用于纤维素酶的改造,并通过这种突变改善了突变体酶分子的某些催化特性。但正如上文所述,通过理性设计对纤维素酶分子进行改造,要求对其三维结构信息要非常清楚,而目前仅有纤维素酶家族(如 GH5-9,GH12,GH45和 GH48) 的少数成员获得了三维结构 (表1);而且,由于目前对纤维素酶的结构与功能关系的理解还非常有限,所以即便在纤维素酶结构已知的情况下对其进行设计,成功的可能性仍难以得到保证,所以至今尚未找到纤维素酶分子改造的普适性方案,也不能完全解析纤维素酶持续性催化过程的分子动态行为。另外,同一家族的纤维素酶分子其序列相似性仅大于30%,这说明同一基因序列在进化过程中发生了一系列位点的替换,但其功能并没有明显改变,这从某种侧面也说明该方法存在低效性问题[23]。正因为如此,对基因进行一系列人为的定点突变时,在筛选过程中并不总能获得功能差异的酶分子。此外,不同纤维素酶间往往具有错综复杂的协同或竞争关系,这都在一定程度上限制了该技术在纤维素酶分子改造上的广泛应用。
2.2 纤维素酶的定向进化
如上所述,由于人们对所要研究的纤维素酶分子结构知之甚少,通过定点突变为代表的理性设计进行纤维素酶分子的改造在短期内仍难以获得广泛的成效。因此,采用“非理性”设计,就成为一种重要的研究手段。所谓“非理性”设计,也即定向进化,就是在对蛋白质的结构及催化机制不是很清楚的条件下,模拟自然进化过程,在体内或体外对基因进行随机突变或体外基因重组,并与高通量筛选策略相结合,最终获得具有某些优良特性的酶分子。该技术首先于1993年由美国科学家Arnold提出;1999年,Michael Himmel指出,非结构信息指导的蛋白质工程策略之一就是定向进化的非理性设计[24]。表3列出了通过定向进化技术成功改变纤维素酶性能的部分实例。
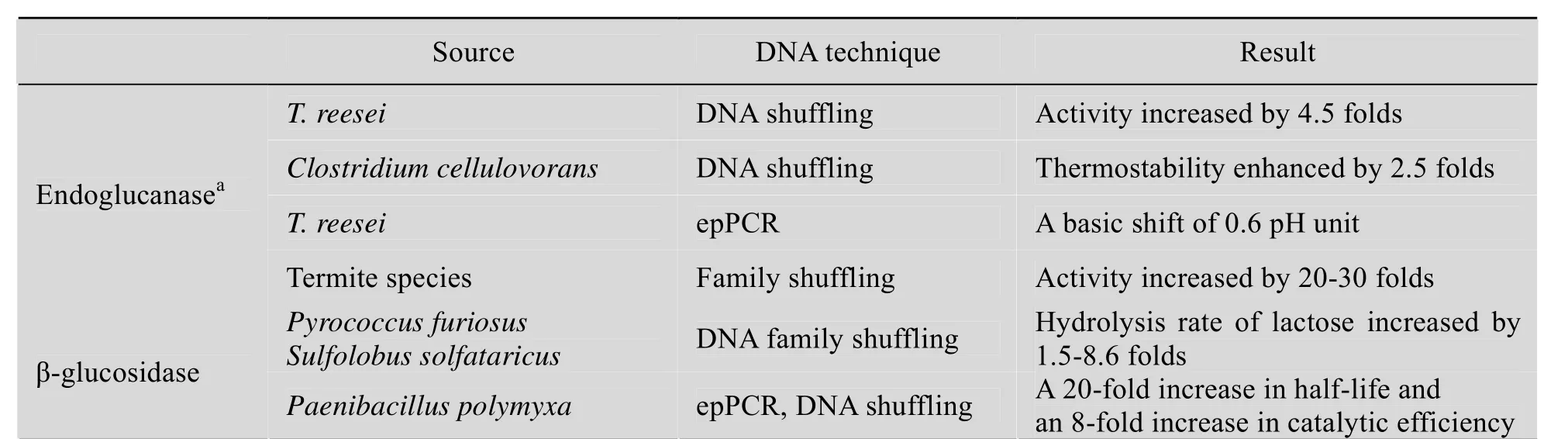
表3 利用定向进化技术完成纤维素酶分子改造的实例*[22]Table 3 List of cellulases whose properties have been changed using directed evolution techniques*[22]
尽管定向进化技术在对纤维素酶进行改造时可能会得到意想不到的“有益收获”,但由于对要改造的蛋白分子结构信息并不清楚,因此操作起来具有明显的“盲目性”。假设一个纤维素酶分子由300个氨基酸组成,其可能的序列空间数目将达20300,而其中有益的突变或组合突变就会非常少,这就使得筛选有效突变体的工作强度非常大,同时也大大降低了其改造的有效性。因此如何构建有效的突变体文库,如何建立合理筛选突变体及正确评价各突变体性能的方法,就成为该策略亟待解决的关键问题与限制性瓶颈[24]。现在,通过对氨基酸密码子表的简化及重复饱和突变 (Iterative saturation mutagenesis,ISM) 技术的引入有效地改进了突变体文库的质量,也明显缩短了突变体的筛选时间。尽管对构建突变体文库进行了优化,但由于筛选方法的限制,该策略目前仍仅集中在对内切纤维素酶和 β-葡糖苷酶的改造上,还尚未见到有关对外切纤维素酶分子进行定向进化的文献报道。因此,仅利用定向进化技术改造纤维素酶仍有一定的局限性。针对新型纤维素酶空间结构、催化机制以及更高效的筛选策略仍有待开发与深入探索。
2.3 基于结构生物信息学的纤维素酶结构理性设计
随着后基因组时代的来临,糖苷水解酶数据库中收录的纤维素酶基因序列数目呈现爆炸式增长[10]。组学数据的快速增长推动了后基因组时代基于蛋白质家族或基于结构生物信息学的理性设计新策略的形成,其特点即是结合了理性设计与定向进化的优势,通过一定的计算模拟与预测指导纤维素酶的改造。由于现有纤维素酶家族是基于序列相似性建立起来的,同一家族不同序列通常会共有一种拓扑空间结构[25]。这样通过对目标蛋白分子所在家族的相关基因进行多序列比对,就可以绘制出其蛋白质家族的特征序列谱及其序列演化关系,再利用该家族已有结构信息就可以快速找出序列谱中的功能氨基酸及其空间组合的变化规律。已有研究表明,这些功能残基及其组合——功能区 (Sector) 在进化上具有很强的相关性,能反映蛋白质的进化历程[26-27]。因此,这种基于蛋白质结构指导的生物信息统计分析,可以大大减小对有益突变空间的搜索强度和范围,增大理性设计成功的概率。另外再辅助以计算机模拟,就可以为更好地探讨纤维素酶与底物的结合及其催化断键机制提供良好的研究平台[28-29],同时能更清晰地绘制出纤维素酶催化过程的反应坐标[18,30],为创造具有新性能或新功能的纤维素酶分子奠定技术基础。
目前,针对蛋白质空间结构优化的理性设计算法已经相继推出,较突出的是 SCHEMA,ProSAR及 ROSETTA[31]。2009年,Heinzelman等基于结构分析,以 3种真菌(特异腐质霉Humicola insolens,嗜热毛壳菌Chaetomium thermophilum和红褐肉座菌H. jecorina)来源的外切纤维素酶 (CBH) Ⅱ超二级结构为单元,进行重组构建嵌合体文库。通过线性回归模型-打分系统计算了每个超二级结构单元对嵌合体稳定性的影响,最终从 38个重组体文库中筛选出了73个突变体,其中15个嵌合体的热稳定性得到了提高 (最高的热稳定性约比热稳定性最强的亲本提高了7 )℃[32-33]。这种基于结构的SCHEMA重组方法比基于序列的 DNA改组(DNA shuffling) 具有更高的效率,同时可以有效地避免重组时的结构坍塌,利于快速筛选出稳定性或其他性能得到提高或改善的突变体。
另外,对于同一家族的序列,可通过重建其祖先基因 (Ancestral sequence reconstruction, ASR)的方法研究其功能的演化过程。通过对同一家族序列的大规模比对分析,引入适当的历史突变;研究对其功能分歧有影响的序列变化,进而重现进化过程中的突变轨迹及其进化关系,该方法现已成功应用于分析类绿色荧光蛋白、类固醇受体及视蛋白的结构、功能与进化的潜在决定因素[15]。
本实验室通过对糖苷水解酶GH5、GH12等一系列家族的纤维素酶序列、结构进行大规模的统计分析,构建了其序列进化树及酶活性中心序列谱。通过对GH12家族序列谱分析后发现,该家族活性部位中含有绝对保守的氨基酸残基(图 2),也有相对保守的氨基酸残基。通过对瑞氏木霉T. reesei来源的内切纤维素酶Cel12A (EG III) 活性部位中绝对保守的 22位色氨酸(Tryptophan,W) 进行定点突变,发现酶分子的活性随突变残基侧链基团π电子的大小而呈现有规律的变化,这与理论计算结果是一致的 (相关结果尚未发表)。这种基于结构生物信息学的统计分析为进一步对纤维素酶分子进行理性设计奠定了理论基础。

图2 糖苷水解酶GH12家族纤维素酶活性部位序列谱中的绝对保守氨基酸统计 (位点编号以瑞氏木霉的Cel12A为参照)Fig. 2 Statistics on the absolutely conservative amino acids in the sequence profile of the active site in GH12 family (Residues are numbered according to Cel12A of T. reesei).
随着CAZy数据库中越来越多的纤维素酶基因序列及结构数据的不断解析,通过对家族内序列的大规模比对及 ASR方法就可重构纤维素酶的祖先基因。利用已有结构进行适当模建,然后引入适当的历史突变,以研究纤维素酶的进化历史和进化轨迹,从而可为纤维素酶活性架构的分析、精确反应坐标绘制及其持续性降解机制的阐释奠定一定的基础;再利用SCHEMA等计算方法就可在一定程度上模拟预测突变体蛋白的结构信息。这种以数据驱动和结构生物信息学指导的纤维素酶结构理性设计方法将会大大加速纤维素酶分子改造的速率,并将成为后基因组时代中对纤维素酶或其他有应用性价值的酶进行改造的新思路。
3 结语与展望
通过蛋白质工程手段对现有纤维素酶分子进行改造,以提高其对结晶纤维素的降解活力或其持续降解性是目前生物燃料工艺亟待解决的关键问题[34]。尽管在过去的几十年中,利用定点突变与定向进化技术在研究纤维素酶的催化机制与改进其催化特性方面都取得了很大的进步,但至今尚未得出提高纤维素酶活力的普遍性规律;对可否持续性提高酶分子的催化效率仍难以给出合理的回答[35]。因此如仅沿用固有思路对纤维素酶进行改造,要达到其工业生产需求的目标还将会经历很长一段时间[36]。
随着基因组学、蛋白质组学、结构组学等技术的迅速发展,以蛋白家族的序列比对与结构模建为基础,通过分析纤维素酶序列、结构与功能的进化关系,研究酶分子的活性架构及其持续性催化的分子动态过程,有助于阐明其催化机制,从而奠定理性设计的理论基础。因此,数据驱动设计与结构生物信息学指导的纤维素酶结构理性设计必将成为后期分子改造的主流方向,也将会为进一步满足生物能源快速发展的需求提供一定的可能。
[1]Ragauskas AJ, Williams CK, Davison BH, et al.The path forward for biofuels and biomaterials.Science, 2006, 311(5760): 484−489.
[2]Himmel ME, Ding SY, Johnson DK, et al. Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science, 2007, 315(5813):804−807.
[3]Wilson DB. Cellulases and biofuels. Curr Opin Biotechnol, 2009, 20(3): 295−299.
[4]Wang JL, Wang LS, Liu WF, et al. Research Advances on the assembly mode of cellulosomal macromolecular complexes. Prog Biochem Biophys, 2011, 38(1): 28−35 (in Chinese).
王金兰, 王禄山, 刘巍峰, 等. 降解纤维素的“超分子机器”研究进展. 生物化学与生物物理进展,2011, 38(1): 28−35.
[5]Wang L, Zhang Y, Gao P. A novel function for the cellulose binding module of cellobiohydrolase I.Sci China Ser C: Life Sci, 2008, 51(7): 620−629.
[6]Cantarel BL, Coutinho PM, Rancurel C, et al. The Carbohydrate-Active EnZymes database(CAZy): an expert resource for glycogenomics. Nucleic Acids Res, 2009, 37(Database): D233−D238.
[7]Lynd LR, Weimer PJ, Van Zyl WH, et al. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev, 2002,66(3): 506−577.
[8]Michael EH. Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy. New Jersey:Wiley Blackwell, 2010 (in Chinese).
M.E. 希默尔. 生物质抗降解屏障——解构植物细胞壁产生物能. 北京: 化学工业出版社, 2010.
[9]Vladimir Uversky, Irina A. Kataeva. Cellulosome.New York: Nova Science Publishers, Inc., 2011 (in Chinese).
V.乌弗斯凯,I.A.卡塔耶瓦. 纤维素降解的超分子机器——纤维小体. 北京:化学工业出版社,2011.
[10]Xie G, Bruce DC, Challacombe JF, et al. Genome sequence of the cellulolytic gliding bacteriumCytophaga hutchinsonii. Appl Environ Microbiol,2007, 73(11): 3536−3546.
[11]Irwin D, Shin DH, Zhang S, et al. Roles of the catalytic domain and two cellulose binding domains ofThermomonospora fuscaE4 in cellulose hydrolysis. J Bacteriol, 1998, 180(7): 1709−1714.
[12]Zhang Z, Wang Y, Wang L, et al. The combined effects of amino acid substitutions and indels on the evolution of structure within protein families. PLoS ONE, 2010, 5(12): e14316.
[13]Zhang Z, Huang J, Wang Z, et al. Impact of indels on the flanking regions in structural domains. Mol Biol Evol, 2011, 28(1): 291−301.
[14]Zhang Z, Xing C, Wang L, et al. IndelFR: a database of indels in protein structures and their flanking regions. Nucleic Acids Res, 2011, 40(D1):D512−D518.
[15]Harms MJ, Thornton JW. Analyzing protein structure and function using ancestral gene reconstruction. Curr Opin Struct Biol, 2010, 20(3):360−366.
[16]Wang L, Zhang Y, Gao P, et al. Changes in the structural properties and rate of hydrolysis of cotton fibers during extended enzymatic hydrolysis.Biotechnol Bioeng, 2006, 93(3): 443−456.
[17]Himmel ME, Ruth MF, Wyman CE. Cellulase for commodity products from cellulosic biomass. Curr Opin Biotechnol, 1999, 10(4): 358−364.
[18]Li J, Du L, Wang L. Glycosidic-Bond hydrolysis mechanism catalyzed by cellulase Cel7A fromTrichoderma reesei: a comprehensive theoretical study by performing MD, QM, and QM/MM calculations. J Phys Chem B, 2010, 114(46):15261−15268.
[19]Pham XH, Tuteja N. Potent inhibition of DNA unwinding and ATPase activities of pea DNA helicase 45 by DNA-binding agents. Biochem Biophys Res Commun, 2002, 294(2): 334−339.
[20]Browne WR, Feringa BL. Making molecular machines work. Nat Nanotechnol, 2006, 1(1):25−35.
[21]Chaparro-Riggers JF, Polizzi KM, Bommarius AS.Better library design: data-driven protein engineering. Biotechnol J, 2007, 2(2): 180−191.
[22]Qu YB. Lignocellulose Degrading Enzymes and Biorefinery. Beijing: Chemical Industry Press,2011: 121−154 (in Chinese).
曲音波. 木质纤维素降解酶与生物炼制. 北京:化学工业出版社, 2011: 121−154.
[23]Gerlt JA, Babbitt PC. Enzyme(re)design: lessons from natural evolution and computation. Curr Opin Chem Biol, 2009, 13(1): 10−18.
[24]Zhang Y-H P, Himmel ME, Mielenz JR. Outlook for cellulase improvement: screening and selection strategies. Biotechnol Adv, 2006, 24(5):452−481.
[25]Gu J, Bourne PE. Structural Bioinformatics. 2nd.New Jersey: Wiley Blackwell, 2009: 419−432.
[26]Halabi N, Rivoire O, Leibler S, et al. Protein sectors: evolutionary units of three-dimensional structure. Cell, 2009, 138(4): 774−786.
[27]Lichtarge O, Wilkins A. Evolution: a guide to perturb protein engineering strategies. Nat Chem Biol, 2010, 5(8): 526−529.
[28]Yan S, Li T, Yao L. Mutational effects on the catalytic mechanism of cellobiohydrolase Ⅰ fromTrichoderma Reesei. J Phys Chem B, 2011,115(17): 4982−4989.
[29]Li T, Yan S, Yao L. The impact ofTrichoderma reeseiCel7A carbohydrate binding domain mutations on its binding to a cellulose surface: a molecular dynamics free energy study. J Mol Model, 2012, 18(4): 1355−1364.
[30]Wilson DB. Processive and nonprocessive cellulases for biofuel production—lessons from bacterial genomes and structural analysis. Appl Microbiol Biotechnol, 2012, 93(2): 493−502.
[31]Bommarius AS, Blum JK, Abrahamson MJ. Status of protein engineering for biocatalysts: how to design an industrially useful biocatalyst. Curr Opin Chem Biol, 2011, 15(2): 194−200.
[32]Heinzelman P, Snow CD, Smith MA, et al.SCHEMA recombination of a fungal cellulase uncovers a single mutation that contributes markedly to stability. J Biol Chem, 2009, 284(39):26229 −26233.
[33]Heinzelman P, Snow CD, Wu I, et al. A family of thermostable fungal cellulases created by structure-guided recombination. Proc Natl Acad Sci USA, 2009, 106(14): 5610 −5615.
[34]Wen F, Nair NU, Zhao H. Protein engineering in designing tailored enzymes and microorganisms for biofuels production. Curr Opin Biotechnol, 2009,20(4): 412−419.
[35]The V Vuong, Wilson DB. Cellulases: Types and Action, Mechanism and Uses. New York: Nova Science Publishers, Inc., 2011: 277−294.
[36]Arnold FH. Combinatorial and computational challenges for biocatalyst design. Nature, 2001,409(6817): 253−257.
November 21, 2012; Accepted: December 24, 2012
Guanjun Chen. Tel: +86-531-88366202; E-mail: guanjun@sdu.edu.cn
国家重点基础研究发展计划 (973计划) (No. 2011CB707402),国家高技术研究发展计划 (863计划) (No. 2012AA10180402),国家自然科学基金 (No. 31170071),山东省国际科技合作项目计划 (鲁科合字[2011]176号第6项) 资助。
Molecular engineering of cellulase catalytic domain based on glycoside hydrolase family
Xiaomei Zhang, Dandan Li, Lushan Wang, Yue Zhao, and Guanjun Chen
State Key Laboratory of Microbial Technology,College of Life Science,Shandong University,Jinan250100,Shandong,China
Molecular engineering of cellulases can improve enzymatic activity and efficiency. Recently, the Carbohydrate-Active enZYmes Database (CAZy), including glycoside hydrolase (GH) families, has been established with the development of Omics and structural measurement technologies. Molecular engineering based on GH families can obviously decrease the probing space of target sequences and structures, and increase the odds of experimental success.Besides, the study of cellulase active-site architecture paves the way toward the explanation of catalytic mechanism. This review focuses on the main GH families and the latest progresses in molecular engineering of catalytic domain. Based on the combination of analysis of a large amount of data in the same GH family and their conservative active-site architecture information, rational design will be an important direction for molecular engineering and promote the rapid development of the conversion of biomass.
cellulase, glycoside hydrolase family, structural bioinformatics, molecular engineering, rational design
Supported by: National Key Basic Research Program of China (973 Program) (No. 2011CB707402), National High Technology Research and Development Program of China (863 Program) (No. 2012AA10180402), National Natural Science Foundation of China (No. 31170071),Shandong International Science and Technology Cooperation Project (LuKeHeZi [2011]No. 176 in item 6).
(本文责编 陈宏宇)
猜你喜欢
中等数学(2022年1期)2022-06-05
生物信息学(2022年1期)2022-04-01
纺织科技进展(2021年3期)2021-06-09
陶瓷学报(2021年1期)2021-04-13
中等数学(2018年4期)2018-08-01
科技创新导报(2018年1期)2018-05-07
石油化工应用(2018年3期)2018-03-24
中学数学研究(广东)(2018年23期)2018-03-05
初中生世界·九年级(2017年9期)2017-10-13
应用化工(2014年11期)2014-08-16
