
海胆状Au-树脂核壳微球的制备及其催化性能
2013-08-16 12:41刘雪锋
服装学报 2013年1期
王 晶, 刘雪锋, 方 云
(江南大学化学与材料工程学院,江苏无锡214122)
20世纪80年代后期,Haruta[1]发现Au纳米颗粒(Au NPs)可高效率地催化CO低温氧化反应,从而打破了Au没有催化活性的传统观念,激发了人们对 Au催化剂研究的强烈兴趣[2]。Valden 等[3]认为,尺寸为2~3 nm左右的Au NPs具有显著的高催化活性;但是,直接将Au NPs用作液相反应催化剂时,反应结束后难以从体系中回收和分离。为此,人们试图将Au NPs负载于固体载体表面,以便实现催化剂回收及其后续循环使用,所用载体大多数为Fe2O3,TiO2,Al2O3,MgO 和 SiO2等无机氧化物[4-9]。然而,由于Au NPs只是简单地沉积或烧结于载体表面,使用过程中Au NPs易于脱落导致催化活性急剧降低,只能循环使用 2 ~ 6 次[10-13]。此后,Pal等[14]将Au NPs静电吸附在阴离子交换树脂微球表面,获得了Au NPs-树脂核壳微球,尽管可以通过简单过滤就可以分离树脂微球,但是水相反应体系中的无机离子依然会使Au NPs容易从树脂微球表面大量脱落。可见,Au NPs无法牢固地负载于载体表面致使其难以有效地循环重复使用,极大地制约了Au催化剂在液相反应体系中的功能和使用。
文中以AuCl4--树脂为前驱体,以抗坏血酸(VC)[15]为还原剂,通过一步简单的液相化学还原反应,低成本高通量地得到柱状Au颗粒根植于树脂表面的海胆状Au-树脂核壳微球;进而以4-硝基酚(4-NP)还原成4-氨基酚(4-AP)的反应为模型体系,考察了海胆状Au-树脂核壳微球作为催化剂及其可重复循环使用性能。
1 材料与方法
1.1 试剂与仪器
1.1.1 试剂 氯金酸(HAuCl4·4H2O),抗坏血酸(VC),Cl型717 阴离子交换树脂,NaBH4,4-NP,均为分析纯,皆由中国医药集团上海化学试剂有限公司提供;4-AP,分析纯,阿拉丁试剂(上海)有限公司提供;超纯水,实验室自制,电导率17.8 MΩ·cm。
1.1.2 仪器 S-4800场发射扫描电镜(FE-SEM),日本日立公司制造;TU-1901型双光束紫外可见(UV-Vis)分光光谱仪,北京普析通用仪器有限责任公司制造。
1.2 海胆状Au-树脂核壳微球的制备
图1 (a)为海胆状Au-树脂核壳微球的制备过程示意。在室温条件下,处理过的Cl型717阴离子交换树脂(图1(b))首先与HAuCl4水溶液经离子交换得到AuCl4--树脂前驱体(图1(c)),再在轻微振摇状态下,用VC水溶液室温还原并老化12 h后,滤出微球,水洗得到金黄色的海胆状Au-树脂核壳微球(图1(d))。采用FE-SEM观察Au-树脂核壳微球的形貌结构,用XRD检测Au的晶体类型。
1.3 催化性能研究
以浓度为0.1 mol/L的NaOH水溶液为溶剂,分别配制系列浓度的4-NP和4-AP标准溶液,在50℃时用紫外 -可见光度法测定吸光度 A和浓度c(mol/L)标准曲线;取浓度为9.0 ×10-5mol/L的4-NP溶液50 mL和0.65 mol/L的 NaBH4溶液11 mL,于100 mL三口烧瓶中混合均匀后,投入0.5 g海胆状Au-树脂核壳微球(湿),在恒温水浴50℃反应,用紫外-可见分光光度计监测反应进程;反应结束后,滤出海胆状Au-树脂核壳微球催化剂,水洗之后,重复进行催化实验10次,其他反应条件相同,每次反应结束后分析4-NP的转化率用以衡量催化剂的催化活性。

图1 海胆状Au-树脂核壳微球制备过程Fig.1 Fabrication of urchin-like Au-Resin core-shell microspheres
2 结果与讨论
2.1 海胆状Au-树脂核壳微球的结构特征
采用FE-SEM观察Au-树脂核壳微球表面Au的形貌结构和排列特征,具体如图2所示。
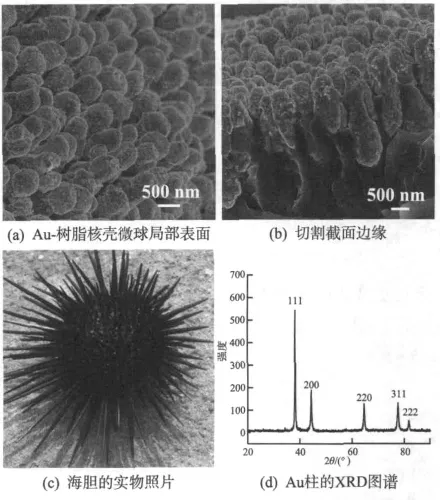
图2 Au-树脂核壳微球结构特征Fig.2 Structural feature of Au-Resin core-shell microspheres
由图2(a)可见,圆柱状Au微柱均匀地呈竖直状态排布在树脂微球的表面;将Au-树脂核壳微球切割后,从其切割边缘(图2(b))可以清晰地观察到,Au微柱的根部深深地植入树脂微球内部,在树脂表面形成单层Au微柱的规则阵列,从而形成以树脂微球为内核、Au微柱单层阵列为外壳的核壳型结构。
Au微柱的长度为3.0 μm左右,其横截直径约440 nm,Au微柱彼此间有100 nm左右的间隙,可见该核壳微球表面的外壳层没有对内核微球表面形成“全封闭”屏障,这种核壳微球与海生动物海胆的形貌非常类似(图2(c)),因此,称之为海胆状Au-树脂核壳微球[16-17]。由于Au微柱的根部植入树脂的可见深度约2.0 μm,可以预见Au微柱与树脂之间结合得非常牢固,有望在后续催化反应中有效地避免Au颗粒从树脂微球表面的脱落问题。
为检验Au微柱与树脂微球之间结合的牢固程度,将所得海胆状Au-树脂核壳微球放置于甲苯中煮沸24 h后用SEM观察,发现Au-树脂核壳微球的外观、其表面Au微柱阵列没有发生任何改变。可见,若是将文中所得海胆状Au-树脂核壳微球用作液相反应催化剂,应当可以有效地避免催化剂的流失问题。最后,用新配制的铬酸洗液蚀除树脂微球,离心得到Au颗粒测试其XRD图谱(图2(d)),与标准卡片(PDF 65-2870)对照,Au颗粒属于面心立方结构[18]。
2.2 海胆状Au-树脂核壳微球的催化性能
以4-NP在碱性水溶液被NaBH4还原成4-AP反应为模型体系,考察所得海胆状Au-树脂核壳微球的催化性能,反应方程式如下:
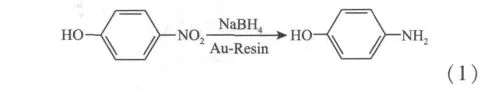
研究表明,若是没有催化剂参与,则该反应不能发生[10-11,19]。
为便于检测反应进程,采用紫外-可见光度法首先测定4-NP和4-AP的标准曲线(见图3)。
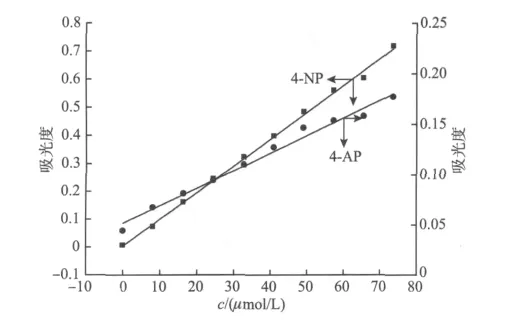
图3 4-NP和4-AP的标准曲线Fig.3 UV adsorption standard curve of 4-NP and 4-AP
经数学拟合得到4-NP和4-AP的标准曲线方程:
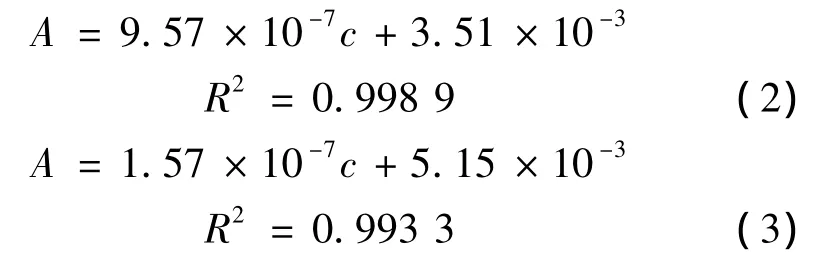
反应过程中,取样用紫外-可见光度计监控反应进程,结果如图4(a)所示。图中在400 nm处为反应物4-NP的吸收峰,317 nm处为产物4-AP的吸收峰;随着反应时间的增加,400 nm处的吸光度A不断降低,而317 nm处的A相应增加,可见海胆状Au-树脂核壳微球具有明显的催化效应。
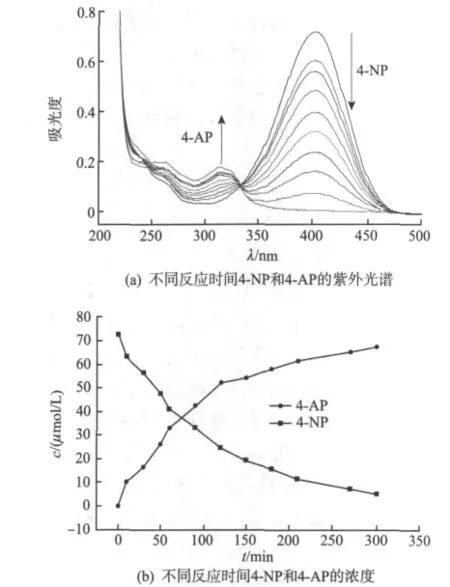
图4 反应过程中4-NP和4-AP的浓度及紫外光谱Fig.4 Concentration and UV adsorption spectrum of 4-NP and 4-AP in the reaction process
图4 (b)是催化反应过程中,体系中4-NP和4-AP的浓度随反应时间t的变化曲线。由图4(b)可以看出,反应时间达到300 min时,体系中4-NP和4-AP的浓度随时间不再有明显变化,所以后续考察催化剂的重复循环使用性能时,将反应时间固定为5 h。由于反应过程中NaBH4的浓度远大于4-NP,将图4(b)中4-NP的浓度数据按照准一级反应拟合,得到线性关系良好的准一级反应速率方程:
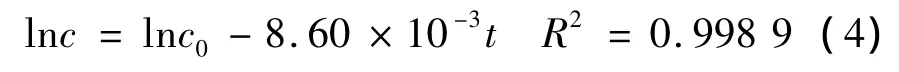
式中,c0为4-NP的起始浓度9.0 ×10-5mol/L,可见该催化反应过程表现出典型的准一级反应特征,其反应速率常数为 8.60 × 10-3min-1。

图5 单颗Au微柱的FE-SEM图Fig.5 FE-SEM images of single Au pillar
文献[3]报道,只有尺寸在纳米范畴的Au NPs才具有明显的催化作用,2~3 nm的Au NPs催化效应最强,随着Au NPs尺寸增加,其催化性能急剧衰减而消失;而文中所得Au微柱的尺寸明显偏大。为阐明其具有催化作用的原因,采用高倍FE-SEM仔细观察Au微柱表面的精细结构,结果如图5所示。高倍FE-SEM结果显示,Au微柱表面布满尺寸大小约20 nm不等的Au NPs。由此可见,文中所得海胆状Au-树脂核壳微球具有明显催化活性的原因在于:Au微柱表面具有呈密集方式排布的Au NPs。因此文中所得速率常数数值与文献[10]相比明显偏小的原因是由于文中所得海胆状Au-树脂核壳微球表面Au NPs尺寸偏大,所以其催化活性与2~3 nm的Au NPs相比明显偏小。
2.3 海胆状Au-树脂核壳微球催化剂的可重复使用性能
反应物料浓度和反应温度均保持不变,反应时间固定为300 min,以4-NP的转化率评价海胆状Au-树脂核壳微球催化剂的可重复利用性能,图6为4-NP的转化率随催化剂重复使用次数的变化关系。由图6可以看出,海胆状Au-树脂核壳微球催化剂重复使用到10次,4-NP的转化率依然保持在85%以上。
由此充分说明,尽管所得Au-树脂核壳微球的催化活性不是十分突出,但却具有良好的稳定性和重复循环使用性能。这与Au-树脂核壳微球的海胆状构造密切相关,Au微柱表面密集的Au NPs(大小约20 nm不等)为催化剂提供催化活性中心;根植深度占其总长度约2/3的Au微柱与树脂微球之间形成非常牢固的负载作用,很好地解决了催化剂的磨损、脱落等流失问题;类似于海胆表面刺状结构的Au微柱呈阵列型排布,Au微柱之间存在着足够的空间,再加上阴离子交换树脂本身对反应体系中阴离子反应物的富集作用,均有利于Au-树脂核壳微球催化作用的发挥。可见,文中研究结果为解决新兴Au纳米负载型催化剂的重复循环使用性能较差的问题提供了很好的思路。
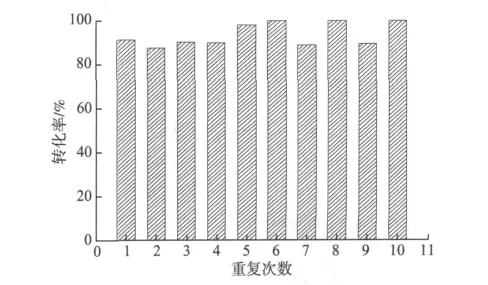
图6 4-NP转化率随催化剂重复使用次数的变化关系Fig.6 Conversion rate of 4-NP vs recycle number of Au-Resin catalyst
3 结语
[1]Haruta M.Catalysis:gold rush[J].Nature,2005,437:1098-1099.
[2]Hutchings G J.New directions in gold catalysis[J].Gold Bull,2004,37(1-2):3-11.
[3]Valden M,LAI X,Goodman D W.Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties[J].Science,1998,281(5383):1647-1650.
[4]Haruta M,Tsubota S,Kobayashi T,et al.Low-temperature oxidation of CO over gold supported on TiO2,α-Fe3O4and Co3O4[J].J Catal,1993,144(1):175-192.
[5]Haruta M.Novel catalysis of gold deposited on metal oxides[J].Catalysis Surveys from Japan,1997,1(1):67-73.
[6]Imai H,Daté M,Tsubota S.Preferential oxidation of CO in H2-Rich gas at low temperatures over Au nanoparticles supported on metal oxides[J].Catal Lett,2008,124(1-2):68-73.
[7]Simakova O A,Kusema B T,Campo B C,et al.Structure sensitivity in L-Arabinose oxidation over Au/Al2O3catalysts[J].J Phys Chem C,2011,115(4):1036-1043.
[8]Costa V V,Estrada M,Demidova Y,et al.Gold nanoparticles supported on magnesium oxide as catalysts for the aerobic oxidation of alcohols under alkali-free conditions[J].J Catal,2012,292:148-156.
[9]Chou J,Franklin N R,Baeck S-H,et al.Gas-phase catalysis by micelle derived Au nanoparticles on oxide supports[J].Catal Lett,2004,95(3-4):107-111.
[10]YOU Liang,MAO Yi-wu,GE Jian-ping.Synthesis of stable SiO2@Au-nanoring colloids as recyclable catalysts:galvanic replacement taking place on the surface[J].J Phys Chem C,2012,116(19):10753-10759.
[11]Kuroda K,Ishida T,Haruta M.Reduction of 4-nitrophenol to 4-aminophenol over Au nanoparticles deposited on PMMA[J].Journal of Molecular Catalysis A:Chemical,2009,298:7-11.
[12]GE Jian-ping,ZHANG Qiao,ZHANG Tie-hui,et al.Core-satellite nanocomposite catalysts protected by a porous silica shell:controllable reactivity,high stability,and magnetic recyclability[J].Angew Chem Inter Ed,2008,120(46):9056-9060.
[13]ZHAN Guo-wu,HONG Ying-ling,Mbah V T,et al.Bimetallic Au-Pd/MgO as efficient catalysts for aerobic oxidation of benzylalcohol:a green bio-reducing preparation method[J].Appl Catal A,2012,439-440:179-186.
[14]Praharaj S,Nath S,Ghosh S K,et a1.Immobilization and recovery of Au nanoparticles from anion exchange resin:resin-bound nanoparticle matrix as a catalyst for the reduction of 4-Nitrophenol[J].Langmuir,2004,20(23):9889-9892.
[15]SUN Kai,QIU Jian-xia,LIU Ji-wei,et al.Preparation and characterization of gold nanoparticles using ascorbic acid as reducing agent in reverse micelles[J].J Mater Sci,2009,44(3):754-758.
[16]LI Jiang-ying,XIONG Sheng-lin,PAN Jun,et al.Hydrothermal synthesis and electrochemical properties of urchin-like coreshell copper oxide nanostructures[J].J Phys Chem C,2010,114(21):9645-9650.
[17]Elias J,Lévy-Clément C,Bechelany M,et al.Hollow urchin-like ZnO thin films by electrochemical deposition[J].Adv Mater,2010,22(14):1607-1612.
[18]REN Yue-ping,XU Cheng-cheng,WU Meng-jie,et al.Controlled synthesis of gold nanoflowers assisted by poly(vinylpyrrolidone)-sodium dodecyl sulfate aggregations[J].Colloids Surf A,2011,380(1-3):222-228.
[19]Panigrahi S,Basu S,Pal S,et al.Synthesis and size-selective catalysis by supported gold nanoparticles:study on heterogeneous and homogeneous catalytic process[J].J Phys Chem C,2007,111(12):4596-4605.
猜你喜欢
小猕猴学习画刊(2022年10期)2022-11-01
传感技术学报(2022年8期)2022-10-25
模具制造(2021年9期)2021-11-02
幼儿画刊(2021年9期)2021-09-20
原子与分子物理学报(2021年2期)2021-03-29
电子元件与材料(2018年11期)2019-01-04
材料科学与工程学报(2018年1期)2018-03-15
西安工程大学学报(2016年6期)2017-01-15
材料科学与工程学报(2016年1期)2017-01-15
北方人(2016年17期)2016-09-14
