
新型非甾体类孕酮受体调节剂的合成与表征
2013-08-16 12:41蒲雯雯陈丽萍林建国罗世能夏咏梅
服装学报 2013年1期
姚 君, 蒲雯雯, 陈丽萍, 林建国,罗世能*,, 邱 玲, 夏咏梅
(1.江南大学化学与材料工程学院,江苏无锡214122;2.江苏省原子医学研究所卫生部核医学重点实验室,江苏省分子核医学重点实验室,江苏无锡214063)
孕酮受体(PR)可以作为影像诊断和放射性治疗乳腺癌的重要作用靶点[1-3]。孕酮受体配体(progesterone receptor ligands,PRLs)主要通过孕酮受体(PR)介导而发挥作用[4-5],按其生物活性可分为孕酮受体激动剂、孕酮受体拮抗剂和选择性孕酮受体调节剂[6-7]。尽管在临床上已经存在一些甾体类孕酮受体调节剂药物的应用,但这些药物仍存在一些不足:如选择性差,交叉反应严重,甚至还会带来一些副作用[7-8]。因而发掘新型的孕酮受体配体是很有必要的。
研究[7,9-10]发现非甾体类的孕酮受体配体具有相对较好的优势,Wyeth Pharmaceuticals等[11-13]最近发现了一类Tanaproget系列化合物,其对孕酮受体具有很高的选择性和亲和力,可作为非甾体类孕酮受体激动剂。文中通过对该类结构作进一步的修饰,期望可以改善其药物作用效果。利用生物电子等排体原理,一方面将Tanaproget结构中硫酮基置换为羰基,虽然其生物活性有可能发生转变[14],但同时又有可能增加其与孕酮受体的亲和力[15-16];另一面对于Tanaproget氟代乙基和氟代丙基衍生物,虽然其具有更高的受体亲和力,但是考虑是否也可以同时利用电子等排体原理将卤素氟置换为羟基从而增强药物水溶性或改变药效。对获得的化合物进一步甲基化,从而为该化合物的正电子放射性核素11C标记实验提供基础。
1 材料与方法
1.1 主要试剂和仪器
1.1.1 试剂 5-溴-2-氨基苯乙酮,质量分数为98%,北京依能特科技有限公司提供;烯丙基氯化镁,四氢呋喃溶液,浓度为1.7 mol/L,百灵威科技有限公司提供;N,N-羰基二咪唑,质量分数为97%,四(三苯基膦)钯,质量分数为99.8%,氯磺酰异氰酸酯,质量分数为98%,均由阿拉丁试剂有限公司提供;1-叔丁氧羰基吡咯-2-硼酸,质量分数为98%,石家庄纳司科医药科技有限公司提供;碘甲烷,分析纯,西亚试剂-成都格雷西亚化学技术有限公司提供;薄层层析硅胶,化学纯,青岛海洋化工有限公司提供;薄层层析(TLC)薄板,浙江省台州市路桥四甲生化塑料厂提供;乙酸乙酯、石油醚、四氢呋喃、甲苯、乙醇、碳酸钾、无水硫酸钠、氯化钠、N,N-二甲基甲酰胺,均为分析纯,皆由国药集团化学试剂有限公司提供。
1.1.2 仪器 PLS-1810型低温反应浴槽,日本东京理化器械株式会社制造;KQ2200E型超声波清洗器,昆山超声仪器有限公司制造;85-2型恒温磁力搅拌器,上海司乐仪器有限公司制造;RE52CS-2型旋转蒸发仪,上海亚荣生化仪器厂制造;BS224S型电子天平,Sartorius公司制造;NICOLET NEXUS470型傅里叶变换红外光谱仪,Thermo Electron Corporation制造;DHG-9023A型电热恒温鼓风干燥器,上海精宏实验设备有限公司制造;MP-51745型微量熔点测定仪,日本岛津公司制造;Bruker AM400型核磁共振波谱仪,德国Bruker光谱仪器公司制造;Waters Platform ZMD4000型质谱仪,美国Waters公司制造。
1.2 实验方法
以2-氨基-5-溴苯乙酮为起始原料,经以下反应合成目标产物。各中间体及目标化合物的合成路线如图1所示。
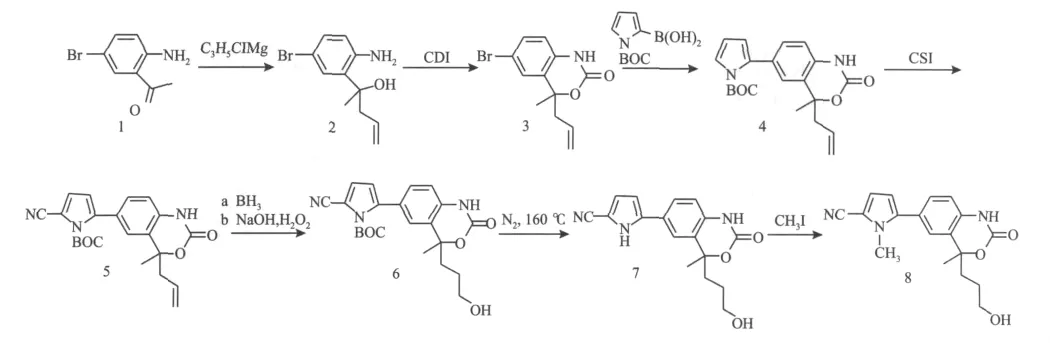
图1 目标产物的合成路线Fig.1 Synthetic route of the target compound
1.2.1 2-(2-氨基-5-溴苯) 戊烷-4-烯-2-醇(2) 的合成 向装有温度计、恒压滴液漏斗和氮气保护下的四口烧瓶中依次加入化合物1(3.00 g,14.01 mmol)和180 mL经钠块重蒸处理过的四氢呋喃溶液,置于 -78℃ 的低温槽中搅拌30 min,再从恒压滴液漏斗逐滴加入烯丙基氯化镁(1.70 mol/L,24.72 mL,42.03 mmol),滴加过程中注意控制滴加速度,始终保持烧瓶内温度维持在-78~-70℃之间,约30 min滴加完毕;在-78℃下搅拌反应5 h后,经TLC(展开剂:V石油醚∶V乙酸乙酯=3∶1)检测判定反应结束;将四口烧瓶取出,待反应液恢复至室温,加入400 mL水,再用50 mL乙酸乙酯萃取3次,有机层用30 mL饱和的氯化钠水溶液洗涤3次,经无水硫酸钠干燥过夜,减压蒸除溶剂,通过硅胶柱层析分离纯化(洗脱液:V正己烷∶V乙酸乙酯=3∶1),得到化合物2黄色油状3.59 g。
1.2.2 1-H-4-甲基 -4-烯丙基 -6-溴苯并[d][1,3]-噁嗪-2(4H)-酮(3)的合成 室温下,将化合物2(3.59 g,14.02 mmol)转移至三口烧瓶中,再加入150 mL经钠块重蒸处理过的四氢呋喃溶液和N,N-羰基二咪唑(CDI,2.50 g,15.42 mmol),在 80℃ 油浴中回流,50 h后经TLC检测反应结束;将反应液冷却至室温,加入300 mL水,用50 mL乙酸乙酯萃取3次,有机层用30 mL饱和氯化钠水洗3次,经无水硫酸钠干燥,减压蒸除溶剂后经硅胶柱层析分离纯化(洗脱液:V石油醚∶V乙酸乙酯=4∶1),得到化合物3棕色色油状物3.09 g。
1.2.3 2-(2-H-1,4-二氢 -4-甲基-4-烯丙基 -2-氧苯并[1,3]噁嗪-6-基)-吡咯-1-羧基酸叔丁基酯(4)的合成 室温氮气保护下,将化合物3(1.55 g,5.49 mmol)溶入50 mL甲苯并转移至250 mL三口烧瓶中,再快速称取四(三苯基膦)钯(162 mg,0.14 mmol)加入烧瓶中,搅拌30 min后,依次加入N-叔丁氧羰基吡咯硼酸(2.32 g,10.98 mmol)的20 mL 乙醇溶液和碳酸钾(1.52 g,10.98 mmol)的20 mL水溶液,之后将三口瓶移至90℃油浴中加热回流,48 h后经TLC检测反应结束;待反应液冷至室温后,将反应液倒入60 mL饱和的碳酸氢钠溶液中搅拌,再用40 mL乙酸乙酯萃取4次,有机相用水和饱和的氯化钠洗涤后经无水硫酸镁干燥,抽滤并减压悬蒸后进行硅胶柱层析分离纯化(V石油醚∶V乙酸乙酯=4∶1),即得到化合物4深棕色油状物1.62 g。
1.2.4 2-(2-H-1,4-二氢 -4-甲基 -4-烯丙基 -2-氧苯并[1,3]噁嗪-6-基)-5-氰基吡咯-1-羧基酸叔丁基酯(5)的合成 在氮气保护下的四口烧瓶中加入化合物4(1.62 g,4.40 mmol)和150 mL 经钠块重蒸处理过得四氢呋喃溶液,置于-78℃低温槽中搅拌,之后用移液管加入氯磺酰异氰酸酯(CSI,47.17 μL,5.28 mmol),反应4 h后经TLC检测原料点基本消失;再加入N,N-二甲基甲酰胺(DMF,2 mL,26.40 mmol)在室温下搅拌1.5 h后,TLC监测反应结束。将反应液倒入400 mL水中搅拌,用50 mL乙酸乙酯萃取3次,有机相用50 mL饱和的氯化铵溶液水洗3次,无水硫酸钠干燥,减压抽滤悬蒸后经硅胶柱层析分离纯化(洗脱液:V石油醚∶V乙酸乙酯=2∶1),得到化合物5白色粉末状固体1.18 g,熔点:123~127℃。
1.2.5 5-(1-H-2,4-二氢 -4-甲基 -4-(3-羟基)-2-氧苯并[d][1,3]噁嗪-6-基)-N-叔丁氧羰基吡咯-2-氰基(6)的合成 将100 mL西林瓶中加入化合物5(420 mg,1.07 mmol)并置于60℃ 真空干燥箱中干燥,2 h后取出并用橡胶塞铝盖密封;再向氮气保护下的西林瓶中加入30 mL事先经钠块重蒸处理过的四氢呋喃溶液,置于 -10℃ 中搅拌,并逐滴加入硼烷四氢呋喃络合物溶液(1.0 mol/L,4.28 mL,4.28 mmol),反应5 h后,经TLC检测原料点消失;然后向西林瓶中滴加氢氧化钠溶液(浓度为4 mol/L,1.07 mL,4.28 mmol),搅拌 15 min 后逐滴加入质量分数30% 双氧水(浓度为10 mol/L,428 μL,4.28 mmol)继续搅拌2 h后,经TLC确证反应结束。将反应液倒入200 mL水中,用30 mL乙酸乙酯萃取4次,有机相用50 mL饱和氯化铵水洗3次,经无水硫酸钠干燥过夜,减压抽滤悬蒸后,经硅胶柱层析分离纯化(洗脱液:V石油醚∶V乙酸乙酯=1∶2),得到化合物6黄色油状物260 mg。
1.2.6 5-(1-H-2,4-二氢 -4-甲基 -4-(3-羟基)-2-氧苯并[d][1,3]噁嗪 -6-基)-1-H-吡咯 -2-氰基(7)的合成 称取化合物6(300 mg,0.73 mmol)置入50 mL单口瓶中,连上真空尾接管,在氮气流的氛围中将单口瓶放入160℃的油浴锅中加热30 min后冷却至室温,得到黏稠的亮黄色油状物。薄层层析分离(展开剂:V二氯甲烷∶V乙酸乙酯=1∶2),得到油状化合物163 mg,即为化合物7。
1.2.7 5-(1-H-2,4-二氢 -4-甲基 -4-(3-羟基)-2-氧苯并[d][1,3]噁嗪 -6-基)-1-甲基 -吡咯 -2-氰基(8)的合成 室温下,依此向50 mL单口瓶中加入化合物7(100 mg,0.32 mmol),DMF(20 mL)溶液和碳酸钾(221 mg,1.60 mmol),搅拌 10 min,加入碘甲烷(20μL,0.32 mmol),5 h后经TLC确定原料点消失。反应结束后向反应器中加入150 mL水,用20 mL乙酸乙酯萃取3次,有机相用20 mL饱和的氯化钠溶液洗涤3次,无水硫酸钠干燥后减压蒸除溶剂,经丙酮充分溶解后,薄层层析分离(展开剂:V二氯甲烷∶V乙酸乙酯=1∶1),得到白色固体化合物70 mg,即为化合物8,熔点175~177℃。
2 结果与讨论
2.1 目标化合物及中间体的波谱表征
目标化合物8及其中间体的波谱表征数据见表1,其中目标化合物8的核磁图谱如图2所示。
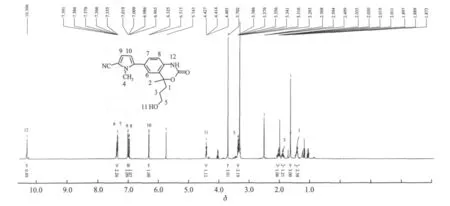
图2 化合物8的1H NMR图谱Fig.2 1H NMR spectrum of compound 8
2.2 反应机理的讨论
因文中实验使用的格氏试剂,四(三苯基膦)钯,N,N-羰基二咪唑,氯磺酰异氰酸酯,硼烷等试剂的特殊性,均要求在尽可能无水无氧氛围中进行,否则会影响反应转化率。反应进程均经TLC(紫外波长254 nm)监测。
2.2.1 格氏反应 格氏加成反应与1.2.5中的硼氢化氧化反应对水分要求较高,所用的溶剂和玻璃仪器均需经无水干燥处理。为了避免反应放热过猛,格氏试剂需逐滴加入,否则会影响反应产率。
2.2.2 N,N-羰基二咪唑的闭环反应 N,N-羰基二咪唑闭环反应实验中,经结构分析N,N-羰基二咪唑的咪唑结构中有一个闭合的大P键且其中一个氮原子上有一对孤对电子,从而决定了CDI具有较强的化学反应活性。尽管如此,但按照等物质的量比投料,原料很难完全反应,因此在实际反应中,通过TLC判断反应进行程度,分批加入CDI,直至反应完全。因CDI及其副产物易溶于水,所以该反应的后处理很方便,产物分离简单,通过对反应液萃取水洗干燥悬蒸后即可直接进入下一步反应中。
2.2.3 Suzuki偶联反应 N-叔丁氧羰基吡咯硼酸与芳环的偶联反应,更倾向于认为其为Suzuki偶联反应[17]。该步反应中,因四(三苯基膦)钯络合物Pd(PPh3)4中Pd配体的厌氧性,故反应必须在氮气等惰性气体下进行[18]。综合文献[19-20]认为该反应经历钯原子被氧化,硼试剂中C-B键异裂,金属钯游离出来络合再生,即氧化加成、硼试剂参与、还原消除3个历程。
2.2.4 氰基取代反应 参照Lohaus方法[21],选用有机碱DMF和CSI将氰基引入吡咯环中[22]。此方法是N-氯磺酰胺在有机碱DMF中分解经历两个循环过渡态转换得到文中所需目标物,即氰基化合物5和副产物DMF·SO3。
2.2.5 吡咯环上氨基的反应 化合物5经硼氢化氧化反应得到端醇化合物6,再经过160℃ 高温氮气保护脱掉吡咯环上的氨基保护基BOC得到化合物7。此方法与三氟乙酸法[23]和硅胶催化法[24]脱除BOC相比,反应时间更短,操作简便,无需进一步处理即可获得纯度较高的产物。所得产物即可投入到下一步甲基化反应中,得到未见报道的经1HNMR确定的新化合物8。在甲基化反应中,考虑到化合物7存在两个较易甲基化的位点(即吡咯环上的氮和酰胺的氮原子),因此,希望可采用加入不同的碱以控制反应的顺利进行。对于苯胺和吡咯类[25-27]的物质,可通过固体碱和溶剂的组合完成甲基化或烷基化反应。在甲基化反应中,固体无机碱可使弱酸性的氨基去质子化,并且固体形式的无机碱所具有的晶格能可以阻止碱作为亲核试剂参与反应;另外,其在反应溶剂中的不溶解可以阻止副反应(如底物分解或直接与碘甲烷反应)的发生[28]。对于甲基化反应,在反应溶剂的选择上,首先排除质子溶剂和能够与强碱反应的溶剂[28],在文献[29-30]中,固体无机碱和DMF的组合更有利于苯胺和吡咯上氮原子的烷基化反应。苯胺上氮的甲基化选用固体无机碱KOH,吡咯环上氮的甲基化选用K2CO3反应[28]。对于化合物7而言,由于右边的氨基,其一边与苯环相连形成p-π共轭,一边与羰基吸电子基相连,要想其发生N烷基化反应,需加入强碱去质子化以形成氮负离子增大其亲核性,而左边吡咯环氮原子的未共用电子对参与杂环上的共轭体系,不易与质子结合,碱性极弱,因而利用碱性极弱的固体碱K2CO3即可使其去质子化发生亲核反应。因此,选用固体无机碱K2CO3和DMF组合可以使化合物7吡咯环上的氮原子更易发生甲基化反应。
3 结语
基于Tanaproget及其衍生物分析,设计合成了一个新型的非甾体类孕酮受体调节剂。利用无机碱K2CO3和DMF组合,可选择性地完成吡咯环上氮原子的甲基化,为后继正电子放射性核素11C标记条件的探索提供了基础和参照。
[1]Benagiano G,Bastianelli C,Farris M.Selective progesterone receptor modulators 3:use in oncology,endocrinology and psychiatry[J].Expert Opin Pharmacother,2008,9(14):2487-2496.
[2]Normanno N,Morabito A,De Luca A.Target-based therapies in breast cancer:current status and future perspectives[J].Endocr Relat Cancer,2009,16(3):675-702.
[3]PENG J,Sengupta S,Jordan V C.Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer[J].Anticancer Agents Med Chem,2009,9(5):481-499.
[4]Mc Ewan I J.Nuclear receptors:one big family[J].Methods Mol Biol,2009,505:3-18.
[5]胡平,任梦鹤,童彩霞,等.非甾体类孕酮受体配体构效关系的研究进展[J].中国新药杂志,2008,17(11):15-20.HU Ping,REN Meng-he,TONG Cai-xia,et al.The research progress of nonsteroidal progesterone receptor ligands structureactivity relationship[J].Chinese Journal of New Drugs,2008,17(11):15-20.(in Chinese)
[6]Winneker R C,Fensome A,Wrobel J E,et al.Nonsteroidal progesterone receptor modulators:structure activity relationships[J].Semin Reprod Med,2005,23(1):46-57.
[7]Winneker R C,Fensome A,ZHANG P,et al.A new generation of progesterone receptor modulators[J].Steroids,2008,73(7):689-701.
[8]江相清,祝丽萍,叶德泳.非甾体类孕酮受体激动剂5-亚苄基-1,2-二氢-5 H-苯并吡喃并[3,4-f]喹啉衍生物三维定量构效关系[J].复旦学报:医学版,2005,32(1):5-9.JIANG Xiang-qing,ZHU Li-ping,YE De-yong.Nonsteroidal progesterone receptor agonist 5-benzal-1,2-dihydro-5H-benzopyran[3,4-f]benzopyranthree-dimensional three-dimensional[J].Journal of Fudan University:Medical Edition,2005,32(1):5-9.(in Chinese)
[9]Pedram B,Van Oeveren A,Mais D E,et al.A tissueselective nonsteroidal progesterone receptor modulator:7,9-difluoro-5-(3-methylcyclohex-2-enyl)-2,2,4-trimethyl-1,2-dihydrochrome no[3,4-f]quinoline[J].J Med Chem,2008,51(13):3696-3699.
[10]ZHANG P,Terefenko E A,Fensome A,et al.6-Aryl-1,4-dihydro-benzo[d][1,3]oxazin-2-ones:a novel class of potent,selective,and orally active nonsteroidal progesterone receptor antagonists[J].J Med Chem,2002,45(20):4379-4382.
[11]Fensome A,Bender R,Chopra R,et al.Synthesis and structure-activity relationship of novel 6-aryl-1,4-dihydrobenzo[d][1,3]oxazine-2-thiones as progester-one receptor modulators leading to the potent and selective nonsteroidal pro-gesterone receptor agonist tanaproget[J].J Med Chem,2005,48(16):5092-5095.
[12]ZHOU Hai-bing,Jae Hak Lee,Christopher G,et al.Imaging progesterone receptor in breast tumors:synthesis and receptor binding affinity of fluoroalkyl-substituted analogues of tanaproget[J].J Med Chem,2010,53(8):3349-3360 .
[13]Jae H L,ZHOU Hai-bing,Carmen S D,et al.Development of[F-18]fluorine-substituted tanaproget as a progesterone receptor imaging agent for positron emission tomography[J].Bioconjugate Chem,2010,21(6):1096-1104.
[14]ZHANG Pu-wen,Terefenko E A,Fensome A,et al.Novel 6-Aryl-1,4-dihydrobenzo[d][1,3]oxazine-2-thiones as potent,selective,and orally active nonsteroidal progesterone receptor agonists[J].Bioorganic and Medicinal Chemistry Letters,2003,13(7):1313-1316.
[15]David J D,Rudiger Faust,Peter J G,et al.Binding affinity and biological activity of oxygen and sulfur isosteres at melatonin receptors as a function of their hydrogen bonding capability[J].Bioorganic Chemistry,2004,32(1):1-12.
[16]Amit Choudhary,Ronald T Raines.An evaluation of peptide-bond isosteres[J].ChemBio Chem,2011,12(12):1801-1807.
[17]王乃兴.有机反应——多氮化物的反应及有关理论问题[M].2版.北京:化学工业出版社,2004:165-171.
[18]Hanhan M E.Ligand effects in palla-dium-catalyzed suzuki and heck coupling reactions[J].Applied Organometallic Chemistry,2008,22(5):270-275.
[19]WANG Nai-xing.Synthesis of 2-Bromo-2'-phenyl-5,5'-thiophene:suzuki reaction versus negishi reaction[J].Synth Common,2003,33(12):2119-2124.
[20]王乃兴.钯催化的交叉偶联反应——2010年诺贝尔化学奖获奖工作介绍[J].有机化学,2011,31(8):1319-1323.WANG Nai-xing.Palladium catalytic cross coupling reaction-thei ntroduction of 2010 nobel prize in chemistry for work[J].Organic Chemistry,2011,31(8):1319-1323.(in Chinese)
[21]Lohaus G.Nitrilsynthesen mit chlorsulfonylisocyanat[J].Chem Ber,1967,100(8):2719-2729.
[22]Hehnut Vorbrtlggen,Konrad Krolikiewicz.The introduction of nitrile-groups into heterocycles and conversion of carboxylic group into their corresponding nitriles with chlorosulfonylisocyanate and triethylamine[J].Tetrahedron,1994,50(22):6549-6558.
[23]Tarikere L G,Narayanan R,Michael J L.Solid-phase synthesis of human mucin-derived O-linked glycopept-ides[J].Letters in Peptide Science,1996,3(2):79-88.
[24]张敏杰,袁修华,马丽,等.硅胶催化的选择性去除N-Boc保护基[J].高等学校化学学报,2007,28(12):2330-2332.ZHAGN Min-jie,YUAN Xiu-hua,MA Li,et al.Silica gel catalytic selective removal N-Boc protecting group[J].Chemical Journal of Chinese Universities,2007,28(12):2330-2332.(in Chinese)
[25]Hayat S A,Rahman M,Choudhary K M,et al.N-Alkylation of anilines,carboxamides and several nitrogen heterocycles using CsFCelite/alkyl halides/CH3CN combination[J].Tetrahedron,2001,57(50):9951-9957.
[26]Makoto O,Akihiro U,Motomitsu Kawai,et al.N-alkylation of aniline derivatives by use of potassium cation-exchanged Y-type zeolite[J].J Chem Soc,Chem Commun,1985,17:1202-1203.
[27]Kavita D,Julien L,Benoit C,et a1.Solvent-promoted and controlled Aza-michael reaction with aromatic amines[J].J Org Chem,2009,74(16):6260-6265.
[28]CAI Li-sheng,XU Rong,GUO Xue-lei,et al.Rapid room temperature11C-Methylation of arylamines with[11C] methylIodide promoted by solid inorganic bases in DMF[J].Eur J Org Chem,2012(7):1303-1310.
[29]Davidson R S,Patel A M,Safdar A,et al.The application of ultrasound to the n-alkylation of amines using phase transfer catalysis[J].Tetrahedron Lett,1983,24(52):5907-5910.
[30]Janusz J,Ryszard O.The application of ultrasound to N-methylation of diazacoronands[J].Tetrahedron Lett,1988,29(8):959-960.
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
昆明医科大学学报(2021年8期)2021-08-13
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
天然产物研究与开发(2016年11期)2016-06-15
合成化学(2015年10期)2016-01-17
应用化工(2014年1期)2014-08-16
华东师范大学学报(自然科学版)(2014年4期)2014-03-11
无机化学学报(2014年4期)2014-02-28
