
双分子荧光互补系统的构建及其在病毒学中的初步应用
2013-07-04 07:23陈春燕韦祖樟袁世山童光志
中国动物传染病学报 2013年3期
张 荣,陈春燕,韦祖樟,袁世山,童光志
(中国农业科学院上海兽医研究所,上海 200241)
病毒在侵入宿主、增殖与传播过程中,其编码的蛋白质是功能发挥的主要载体。研究病毒蛋白之间、或者病毒蛋白与宿主蛋白之间的相互作用,将有助于对病毒致病机制的了解和疾病防控。蛋白质互作研究手段很多,但新发展起来的的双分子荧光互补技术(bimolecular fluorescence complementation,BiFC)[1],因其操作要求简单快捷,可以特异且十分灵敏地检测体内微弱或者瞬时反应的蛋白互作[2-4],能够在生理条件下直接观察单个活细胞内蛋白的相互作用及作用区域,已越来越受到研究人员的青睐。BiFC的基本原理是将荧光蛋白分割成两个失去活性的片段,分别连接目的蛋白,并在细胞内融合表达。若两个目的蛋白存在相互作用而靠拢,两个荧光片段也会被拉近而重新形成有活性的荧光基团,能在荧光显微镜下能直接观察到激发的荧光。鉴于BiFC的优越性,本试验旨在构建BiFC技术平台,为以后深入研究病毒编码蛋白之间及其与宿主蛋白间的相互作用提供便捷。
1 材料与方法
1.1 细胞 质粒与生物试剂非洲绿猴肾细胞系(MARC-145)、含猪繁殖与呼吸综合征病毒(Porcine reproductive and respiratory syndrome virus,PRRSV)核衣壳蛋白(nucleocapsid,N)的质粒pAPRRS由本实验室保存;含黄色荧光蛋白的质粒pEYFP-N1 购自BD-Clontech公司;高保真聚合酶pfuUltraII购自Stratagene公司;限制性核酸内切酶购自NEB公司;禽源反转录酶AMV购自TaKaRa公司;化学转染试剂Fugene HD购自Roche公司;QIAprep® Spin Miniprep Kit质粒提取试剂盒、RNA提取试剂盒购自QIAGEN公司。
1.2 引物设计根据黄色荧光蛋白EYFP和突变体Venus序列差异[5],运用Primer 5.0软件设计一系列突变引物,用于突变EYFP为Venus。同时,设计引物将Venus从第173和174氨基酸位点间分割蛋白为两部分:含N端的VN和含C端的VC。另外,设计引物用于扩增PRRSV的N基因和MARC-145细胞里的α-tubulin基因。引物序列见表1。
1.3 细胞RNA提取为了扩增细胞内α-tubulin基因,将长满单层的MARC-145细胞用QIAGEN公司的RNA提取试剂盒提取细胞内总RNA,然后参照TaKaRa公司AMV反转录酶说明书进行反转录获得cDNA。
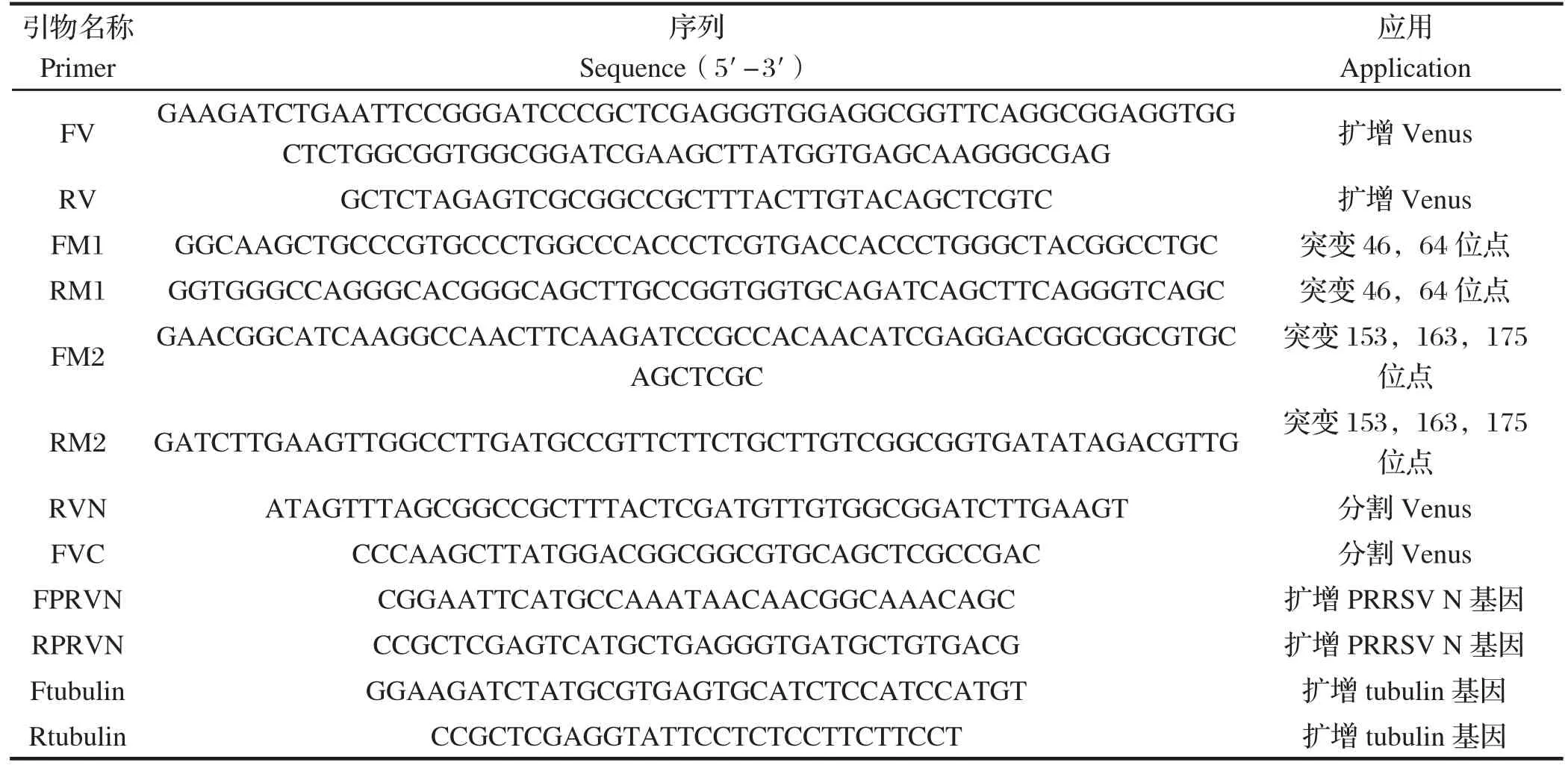
表1 用于构建BiFC质粒的相关引物Table 1 Primer sequences for the construction of BiFC plamids
1.4 PCR与质粒构建以pEYFP-N1为模板,用引物对FV/RM1,FM1/RM2,FM2/RV进行PCR扩增,经琼脂糖凝胶电泳回收目的片段,分别名为S1、S2、S3。再以S1和S2为模板,以引物对FV/RM2扩增,新的PCR融合片段为S12;然后以S12和S3为模板,以FV/RV为引物,将两个片段PCR融合成全长Venus基因。该片段经BglII/NotI酶切后,连入到相应的pEYFP-N1质粒里替换EYFP,新的质粒命名为Venus。接着,再以该Venus为模板,以FV/RVN和FVC/RV为引物,将该蛋白分割为两片段,经BglII/NotI酶切后,连入到Venus质粒里替换全长Venus基因,新的质粒分别命名为VN和VC。为了验证BiFC片段,以pAPRRS质粒为模板,以FPRVN/RPRVN为引物扩增PRRSV的N基因,并经EcoRI/XhoI酶切,分别连入到VN和VC里,获得质粒N-VN和N-VC;以α-tubulin cDNA为模板,以Ftubulin/Rtubulin为引物扩增了tubulin基因,并经BglII/XhoI酶切,分别连入到VN和VC里,获得质粒Tubulin-VN和Tubulin-VC。所有质粒都经过测序鉴定无误。
1.5 质粒转染与荧光检测将构建的质粒经转化大肠杆菌扩大培养后,用 QIAprep® Spin Miniprep Kit提取质粒,并进行浓度测定。将MARC-145细胞接种于6孔细胞板后,待细胞达到约60%密度时,将质粒各取500 ng进行共转染。具体步骤参照Fugene HD试剂说明书执行。待转染16 h后,弃去部分培养基,然后将细胞置于荧光倒置显微镜下观察荧光并拍照保存。
2 结果
2.1 Venus突变体的构建根据突变体Venus与EYFP序列比较结果显示,Venus共存在5个特异的氨基酸位点突变,分别为:F46L、F64L、M153T、V163A、S175G。构建示意图如图1所示。在突变PCR过程中,为便于以后插入目的基因,在其N端分别引物了EcoRI,BamHI和XhoI酶切位点。同时,为防止蛋白空间位阻的影响,在插入目的基因与Venus基因间引入了GGGGS的三个重复序列[6,7](图1)。该突变体Venus和pEYFP-N1质粒经转染细胞16 h后,荧光显微镜观察显示两者均发出绿色荧光,说明突变操作并未影响该蛋白的活性。
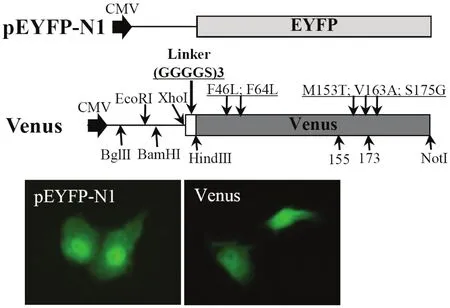
图1 突变体Venus的构建与荧光检测Fig.1 Construction and fl uorescence detection of the Venus mutant
2.2 VN与VC截断片段的构建在突变获得全长Venus基因后,采用PCR将其分割为两部分:含N端173个氨基酸的VN和余下C端的VC。VN和VC质粒均保留了N端的酶切位点及连接序列。两质粒经单独转染和共转染后16 h,甚至延长到32 h,在荧光显微镜下均未能检测到荧光产生(图2),说明Venus在分割为两段后,失去了激发产生荧光的能力;同时也说明该两片段在细胞内共表达后,在没有相互作用的目的蛋白连入时,并不能相互靠拢而形成有活性的荧光基团。
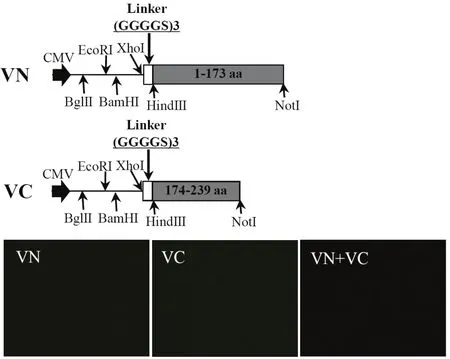
图2 VN和VC片段的构建与荧光检测Fig.2 Construction and fl uorescence detection of the VN and VC fragment
2.3 PRRSV N蛋白的插入与验证PRRSV N蛋白已经通过其他方法证明其可以形成同源二聚体,存在有明显的相互作用[8]。为了进一步验证VN和VC的可行性,我们将PRRSV N基因克隆到VN和VC上,观察两者在共转染时,是否因为N蛋白的相互作用而产生荧光。如图3所示,当N-VN和N-VC单独转染时,并不能产生荧光;而当两者共转染时,产生了明亮的绿色荧光。说明当目的蛋白存在相互作用时,可以将VN和VC片段彼此拉拢而重新形成有活性的荧光基团。
2.4 细胞骨架蛋白α-tubulin的插入与验证上面我们已经证明了VN和VC可以用于检验蛋白的相互作用。但为排除假阳性,我们从用于转染的细胞系MARC-145中扩增出细胞骨架蛋白α-tubulin基因(图4)。该基因全长1353 bp,451个氨基酸,序列比较发现与来源于人和仓鼠的基因完全相同。然后,将该基因连入到VN和VC,共转染PRRSV N蛋白与细胞蛋白α-tubulin,看能否产生荧光。结果显示:N-VN/Tubulin-VC和N-VC/Tubulin-VN没有荧光产生,而Tubulin-VN和Tubulin-VC共转染会产生荧光。该实验说明该相互作用是特异的,排除了假阳性的发生,也进一步证明了该VN和VC用于研究蛋白质相互作用的可靠性。
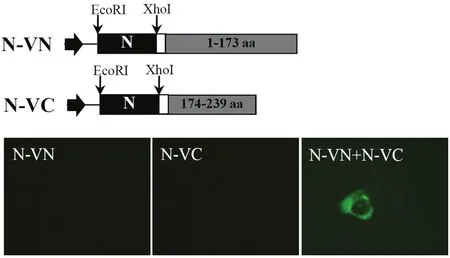
图3 PRRSV N蛋白的插入与荧光检测Fig.3 Construction of PRRSV N inserts and fl uorescence detection
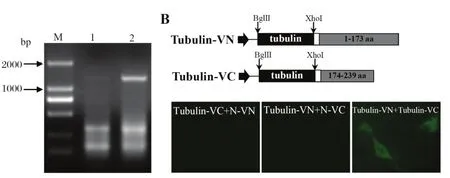
图4 Tubulin基因的扩增(A)及Tubulin-VC、Tubulin-VN构建示意图(B)Fig.4 Amplifi cation of the α-tubulin gene (A) and construction of the α-tubulin inserts for fl uorescence detection (B)
3 讨论
本文通过将黄色荧光蛋白EYFP突变为Venus,然后于第173位氨基酸位点分割成两部分,构建了BiFC片段VN和VC;并分别与已证明存在相互作用的PRRSV N基因连接,在共转染的活细胞内观察到了荧光;同时,连入了细胞骨架蛋白α-tubulin来排除其假阳性。结果显示该技术用于研究蛋白互作是可行的。
BiFC的互补片段来源可以采用EGFP、EYFP,ECFP等[2],但因其片段互补产生有活性的荧光基团受温度、成熟时间、发光强度等因素制约。目前证实采用EYFP的两个突变体Citrine和Venus构建的荧光片段可在37℃生理条件下形成稳定生色团,尤其Venus形成荧光强度更高[7]。因此,本实验选用了Venus构建BiFC系统。
在选择两片段的分割位点上,亦有不同组合[1,2,7,9]:在第154氨基酸位点分割成两段;在第173位点分割成两段;也有研究人员选择N端173氨基酸和从156氨基酸为起点的C端,这样两片段有20多位氨基酸的重叠片段,有利于荧光基团的生成。本实验在最初选择分割位点时,采用了后者的有部分重叠策略。但当两片段共转染时发现有很强的荧光背景(结果未显示)。故我们重新选择了从第173位点直接分割成两部分,当两者共转染时,产生的荧光背景十分微弱(图2),说明选择该分割位点更为可靠。我们在构建该系统时,还在片段的N端引入了四个酶切位点,以方便我们在以后使用时插入目的蛋白。
BiFC的一个优点是高度的灵敏性,但也往往会造成试验结果的假阳性。为了进一步验证该技术是可靠实用的,我们克隆了细胞骨架蛋白α-tubulin,并与PRRSV病毒的N蛋白共转染,发现当两个不相关的蛋白在细胞中共表达时没有荧光产生,这将有利于排除因非特异性结合反应而导致的假阳性;在共转染过程中,我们控制在六孔板中每个质粒的转染量不超过 500 ng,同时,在转染后 12~16 h 即观察荧光;另外,我们还删除了存在于pEYFP-N1中的Kozak序列,以降低蛋白的表达水平。这些手段将减少因蛋白高量表达而造成的假阳性发生。为了排除目的蛋白和Venus片段的空间位阻效应而导致假阴性,我们在两者间引入了一段疏水和亲水氨基酸分隔排列的重复序列,以保持两端蛋白的线性连接,减小了蛋白空间结构的影响。
自BiFC技术建立以来,因其有如常规的免疫共沉淀等体外方法不可比拟的优越特性,已在生物各个领域里得到了广泛的采用。例如在病毒学中,运用该方法研究编码蛋白与宿主的相互作用、基因组复制转录与病毒颗粒包装等[10-17]。但BiFC作为有力的实验工具,也有许多限制。例如荧光片段互补连接后的不可逆性限制了蛋白互作的动态可逆过程研究。因其高度灵敏性,使用过程中得小心区分非特异的背景信号,以免造成假阳性。同时,如有不可避免的蛋白空间位阻效应,也可能产生假阴性。故在实际使用过程中,应与其他多种研究蛋白互作方法结合起来验证。目前,BiFC已发展了多色荧光互补技术(multicolor fluorescence complementation,MFC),将多种荧光蛋白分割后进行组合,可以用于同时检测多种蛋白质的相互作用[2]。总之,BiFC存在诸多限制,但自身还在不断发展和完善,相信会在生物学领域得到更广泛的应用。
[1]Hu C D, Chinenov Y, Kerppola T K.Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation[J].Mol Cell, 2002, 9(4): 789-798.
[2]Hu C D, Kerppola T K.Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis[J].Nat Biotechnol, 2003, 21(5): 539-545.
[3]Kerppola T K.Complementary methods for studies of protein interactions in living cells[J].Nat Methods, 2006,3(12): 969-971.
[4]Shyu Y J, Hu C D.Fluorescence complementation:an emerging tool for biological research[J].Trends Biotechnol, 2008, 26(11): 622-630.
[5]Nagai T, Ibata K, Park E S,et al.A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications[J].Nat Biotechnol, 2002,20(1): 87-90.
[6]Shimozono S and Miyawaki A.Engineering FRET Constructs Using CFP and YFP[M].Methods in Cell Biology, 2008, (85): 381-393.
[7]Shyu Y J, Liu H, Deng X,et al.Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions[J].Biotechniques, 2006, 40(1): 61-66.
[8]Wootton S K, and Yoo D.Homo-oligomerization of the porcine reproductive and respiratory syndrome virus nucleocapsid protein and the role of disulfide linkages[J].J Virol, 2003, 77(8): 4546-4557.
[9]Walter M, Chaban C, Schutze K,et al.Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation[J].Plant J, 2004, 40(3):428-438.
[10]Aparicio F, Sanchez-Navarro J A and Pallas V.In vitro and in vivo mapping of the Prunus necrotic ringspot virus coat protein C-terminal dimerization domain by bimolecular fluorescence complementation[J].J Gen Virol, 2006, 87(Pt 6): 1745-1750.
[11]Atanasiu D, Whitbeck J C, Cairns T M,et al.Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion[J].Proc Natl Acad Sci USA, 2007, 104(47):18718-18723.
[12]Jin J, Sturgeon T, Chen C,et al.Distinct intracellular trafficking of equine infectious anemia virus and human immunodeficiency virus type 1 Gag during viral assembly and budding revealed by bimolecular fluorescence complementation assays[J].J Virol, 2007, 81(20): 11226-11235.
[13]Kenney S P, Lochmann T L, Schmid C L,et al.Intermolecular interactions between retroviral Gag proteins in the nucleus[J].J Virol, 2008, 82(2): 683-691.
[14]Hemerka J N, Wang D, Weng Y,et al.Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay[J].J Virol, 2009, 83(8): 3944-3955.
[15]Zamoto-Niikura A, Terasaki K, Ikegami T,et al.Rift valley fever virus L protein forms a biologically active oligomer[J].J Virol, 2009, 83(24): 12779-12789.
猜你喜欢
中国现代医学杂志(2022年16期)2022-08-31
成都医学院学报(2022年4期)2022-08-19
亚热带农业研究(2022年1期)2022-08-08
实用医学杂志(2022年11期)2022-07-18
江西农业学报(2021年4期)2021-04-20
农业科技通讯(2021年1期)2021-03-06
三农资讯半月报(2020年11期)2020-06-21
中国预防兽医学报(2020年1期)2020-06-05
中国农业科技导报(2020年3期)2020-03-15
中国实验诊断学(2019年11期)2019-11-26
