
离子阱质谱分析仪在研究气态配位化合物形成及配位平衡方面的应用
2013-05-28 07:23杨鹏翔徐锐锋刘顺英胡文浩
质谱学报 2013年1期
杨鹏翔,徐锐锋,赵 芸,刘顺英,胡文浩
(1.华东师范大学上海分子治疗与新药创制工程技术研究中心,上海 200062;2.中国计量科学研究院,北京 100013)
近年来,质谱技术的快速发展及其在生物、无机和有机金属化学领域的广泛使用极大地推动了质谱分析仪在气相反应平衡研究中的应用[1-4]。在质谱分析中,由 Cooks教授最先提出的动力学方法已被用于气态热动力化学的测定,包括气相酸度、碱度、质子亲和力、电子亲和势、电离能、金属离子的亲合力,以及配位分子的空间异构电场对阳离子的亲合力、能量分配和手性分析上的作用[3-8]。该理论将离 子-分 子反应(I-M反应)和碰撞诱导解离(CID)技术相结合,用以表征中心离子配位数及中心离子和配体之间的络合[9-12]。螯合配体和溶剂配体与过渡金属离子的气相I-M反应的平衡常数也已被用于确定配位键的强弱[13-17]。Vachet等研究了过渡金属离子和配合物之间的I-M反应,发现配位数为3的络合体系很容易与溶液试剂(包括水和甲醇)发生络合反应[11]。
本研究利用质谱分析仪的CID功能产生可控的外界能量使原有的过渡金属配合物的配位键断裂,同时利用质谱分析过程中的I-M反应研究络合体系的重新建立和平衡。通过对配位化合物[(Cys-H)CoII(MeOH)]+(配位数为4)在质谱离子阱中的动力学行为分析发现:CID可以使甲醇和CoII之间的配位键断裂,破坏初始的络合平衡体系,致使络合体系的配位数由4降为3。新形成配位数为3的络合体系在CID的激发下,通过I-M反应捕获配体H2O,形成新配位键,得到配位数为4的配位化合物[(Cys-H)CoII(H2O)]+;之后,该配位化合物在CID的冲击下失去配体H2O,随即又捕获MeOH分子回到初始的配位数为4的化合物[(Cys-H)CoII(MeOH)]+。以上过程简而言之,稳定的高配位数化合物在CID的冲击下变成不稳定的低配位数化合物,随后通过I-M反应捕获新的配体,返回能量更稳定的高配位数化合物,重新达到平衡。这种配位平衡的打破和新平衡的自动建立包括配体损失、捕获和交换等步骤。结合CID和I-M反应,可以利用离子阱质谱仪研究在气相中的配位化合物的形成,配位新平衡的建立,并比较配位化合物的稳定性以及可控的配位平衡等。
1 试验部分
1.1 仪器与试剂
Thermo LCQ离子阱质谱系统:美国Thermo Fisher公司产品,配有电喷雾离子源(ESI)及Xcalibur1.2数据处理系统。
所有化学品均为 Aldrich Chemical(St.Louis,MO,USA)公司产品,使用前并未进行纯化处理;所有溶剂均为色谱纯:Aldrich Chemical公司产品;纯水由超纯水系统制备(超纯水系统为美国Millipore公司产品)。
1.2 配位化合物溶液的配制
在样品准备时,半胱氨酸和氯化钴溶解在V(甲醇)∶V(水)=50∶50的混合溶液中,最终获得氨基酸浓度为5×10-3mol/L,金属盐浓度为5×10-4mol/L。
1.3 质谱条件
电喷雾离子源正离子检测模式(ESI+),喷雾电压4.5kV,毛细管电压15V,毛细管加热温度150℃,鞘气(N2)流量40units;使用直接进样的方法以10μL/min的流速进样;所有涉及CID实验离子选择隔离宽度为m/z4,激活Q值为0.25;所有质谱是50个连续扫描的平均。
2 结果与讨论
2.1 配位数与仪器参数设置的关系
配位化合物的形成以及它们之间的相对比例与ESI源的参数设置有关。含5×10-3mol/L半胱氨酸和5×10-4mol/L氯化钴的V(甲醇)∶V(水)=50∶50的混合溶液以10μL/min的流速进入离子阱质谱仪ESI源,其质谱图示于图 1a。其 中,m/z211 为 钴 配 位 化 合 物[(Cys-H)CoII(MeOH)]+,m/z122为半胱氨酸离子[Cys+H]+,m/z140为半胱氨酸水合离子[Cys+H2O+H]+。当毛细管的温度从150℃降至100℃后,在ESI源中有更多的钴离子配位化合物生成,同时还有配位数更高的钴离子配位化合物生成,如[(Cys-H)CoII(MeOH)(H2O)]+及[(Cys-H)CoII(MeOH)2]+,示于图1b。该实验表明,电喷雾电离源(ESI)可以用来产生气态过渡金属离子配合物,并可以通过改变仪器参数在一定程度上控制配位数的高低。

图1 含有5×10-3 mol/L半胱氨酸和5×10-4 mol/L氯化钴的V(甲醇)∶V(水)=50∶50混合溶液的ESI+质谱图Fig.1 ESI mass spectrum of V(methanol)∶V(water)=50∶50 solution containing CoCl2(5×10-4 mol/L)and cysteine(5×10-3 mol/L)in positive mode
2.2 不同配位化合物在CID源作用下的相互转化
离子阱质谱分析仪的CID可以打断配位化合物的配位键。m/z211的[(Cys-H)CoII(MeOH)]+在离子阱CID的作用下,中心离子CoII与配体MeOH的配位键被打断,失去一分子的 MeOH后成为m/z179[(Cys-H)CoII]+;随后捕获H2O生成新的m/z197的配位化合物[(Cys-H)CoII(H2O)]+,示于图2a。m/z179的[(Cys-H)CoII]+在CID的作用下可以捕获配体MeOH和H2O,变成高配位数的配位化合物[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+,示 于 图 2b。[(Cys- H)CoII(H2O)]+在离子阱中同样会在CID的作用下遵循“失去配体,同时捕获新配体”这一规律,分别生成 [(Cys- H)CoII]+和 [(Cys- H)CoII(MeOH)]+,示于图2c。
3个配位化合物 [(Cys-H)CoII(MeOH)]+,[(Cys-H)CoII(H2O)]+和[(Cys-H)CoII]+的相互转化可以在MS2~MS6连续CID过程中观察到,示于图3。由于每次CID都会有部分离子损失,离子数量在MS6后太少,已无法检测。可以推测,如果没有离子损失,在CID的激发下这种配位化合物之间的转化将不断进行。
这3个配位化合物之所以能够相互转化,究其原因是由配位化合物的结构及离子阱中的气态环境决定的。对于配位化合物[(Cys-H)CoII]+,半胱氨酸和钴离子之间有3个配位键[11-12,18-19],配位数为3,该结构是一种不饱和的配位结构,而配位数为4的钴离子配位化合物更加稳定[20],示于图4。[(Cys-H)CoII]+倾向于捕获另外一个配体达到更稳定的结构,如图4中的[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+。MeOH和H2O作为样品的溶剂和样品一起通过ESI气化变成气态分子,持续地进入离子阱质谱仪。因此,离子阱中持续地存在着MeOH和H2O分子,它们作为配体参与了钴离子配位化合物地形成和转化。

图2 30ms CID激发下的MS/MS质谱图Fig.2 MS/MS spectrum of the cysteine-bound cobalt(II)complex under CID with 30ms collision time
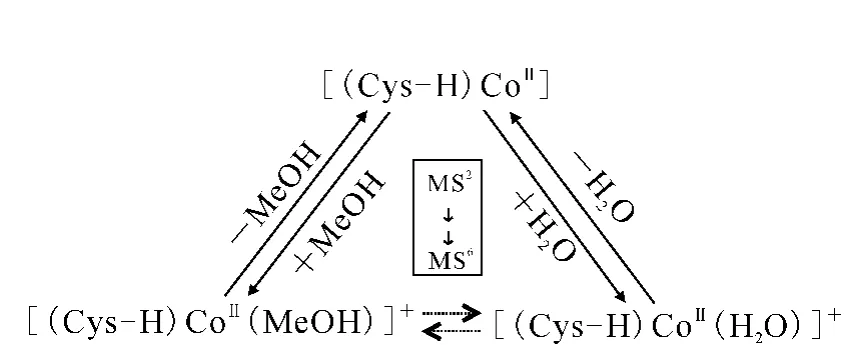
图3 3个半胱氨酸钴(Ⅱ)配合物在多级CID(MS2~MS6)激发下的相互转化关系图Fig.3 The scheme of the transformation among the cysteine-bound cobalt(II)complexes in the course of MSn(n=2to 6)
2.3 CID激发时间与3个配位化合物在离子阱中相对丰度之间的关系
在使用CID技术打断配位键时,可以通过控制CID的激发时间来控制I-M的反应时间。在[(Cys-H)CoII(MeOH)]+的CID实验中,当CID刚开始作用时,[(Cys-H)CoII(MeOH)]+立即失去 MeOH 产生大量的[(Cys-H)CoII]+,示于图5。随着CID激发时间的增加,[(Cys-H)CoII]+的数量逐渐减少,同时[(Cys-H)CoII(H2O)]+出现并逐渐增多。这说明[(Cys-H)CoII]+捕获了离子阱中的 H2O分子,生成[(Cys-H)CoII(H2O)]+[11]。随着 CID激发时间的增加,[(Cys-H)CoII(MeOH)]+减少的速度越来越慢,200ms后出现反弹。说明[(Cys-H)CoII]+捕获了离子阱中的 MeOH 分子,从而回到了[(Cys-H)CoII(MeOH)]+的结构。同时,200ms后[(Cys-H)CoII(H2O)]+也减缓了增长速度,700ms后转向下降。这说明在生成[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+之间有一定的竞争。在CID激发时间1 500ms时,[(Cys-H)CoII(H2O)]+依然在下降,而[(Cys-H)CoII(MeOH)]+依然在上升。
由于LCQ的离子阱最长只能激发1 500ms的CID,因此无法知道最终平衡时,3个配位化合物在离子阱的相对丰度。而[(Cys-H)CoII(H2O)]+的CID实验可以提供线索,比较这3个配位化合物的稳定性。在该实验中,[(Cys-H)CoII(H2O)]+在 CID 的激发下立即失去H2O生成[(Cys-H)CoII]+,生成的[(Cys-H)CoII]+会捕捉离子阱中的H2O和 MeOH,变成更稳定的高配位数离子[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+,示于图6。在CID的800ms激发时间后,[(Cys-H)CoII(MeOH)]+的量已经超过[(Cys-H)CoII(H2O)]+,并且在1 500ms的 CID 激发时,[(Cys-H)CoII(MeOH)]+的增加依然是直线上升,而且[(Cys-H)CoII(H2O)]+和[(Cys-H)CoII]+仍是持续减少。这意味着在LCQ离子阱中更倾向于生成[(Cys-H)CoII(MeOH)]+,这一结论也可以在[(Cys-H)CoII]+的CID试验中得到验证。
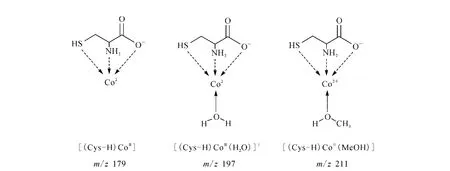
图4 半胱氨酸钴(Ⅱ)配合化合物的结构Fig.4 The structures of the cysteine-bound cobalt(II)complexes

图5 在不同的CID激发时间下,配位化合物离子[(Cys-H)CoII(MeOH)]+和其失去配体 MeOH后的[(Cys-H)CoII]+及再次捕获配体H2O后生成的[(Cys-H)CoII(H2O)]+ 三者之间丰度的变化Fig.5 Under the varied CID collision time to the target complex[(Cys-H)CoII(MeOH)]+ ,he fraction of ion abundance from [(Cys-H)CoII]+ ,[(Cys-H)CoII(H2O)]+ ,and[(Cys-H)CoII(MeOH)]+

图6 在不同的CID激发时间下,配位化合物离子[(Cys-H)CoII(H2O)]+和其失去配体 H2O后的[(Cys-H)CoII]+及再次捕获配体MeOH后生成的[(Cys-H)CoII(MeOH)]+三者之间丰度的变化Fig.6 Under the varied CID collision time to the target complex[(Cys-H)CoII(H2O)]+ ,the fraction of ion abundance from[(Cys-H)CoII]+,[(Cys-H)CoII(H2O)]+and[(Cys-H)CoII(MeOH)]+
在离子阱中对[(Cys-H)CoII]+施加CID时,[(Cys-H)CoII]+并没有解离为更小的离子,反而从一起进入离子阱的溶剂分子中捕获一个配体,生成配位数为4的[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+,示于图7。虽然最初[(Cys-H)CoII(H2O)]+多于[(Cys-H)CoII(MeOH)]+,但在 CID 为1 100 ms的激发时间后,[(Cys-H)CoII(MeOH)]+的量超过[(Cys-H)CoII(H2O)]+并持续直线增加。因为LCQ质谱分析仪是用涡轮泵(turbo pump)抽走气态分子的,而涡轮泵的效率与分子质量成反比,所以离子阱中H2O的量远大于MeOH。因此,在[(Cys-H)CoII]+受到外来能量激发的初始阶段,其与配体H2O反应生成[(Cys-H)CoII(H2O)]+的反应速率较大,这一阶段,以H2O为配体的化合物为主要产物。与H2O相比,MeOH是更强的路易斯碱 (lewis basic),更容易提供电子对给不饱和配位化合物[11],即[(Cys-H)CoII(MeOH)]+的稳定性高于[(Cys-H)CoII(H2O)]+。因此,随着反应地进行,越来越多的[(Cys-H)CoII]+捕获配体MeOH生成更加稳定的[(Cys-H)CoII(MeOH)]+。
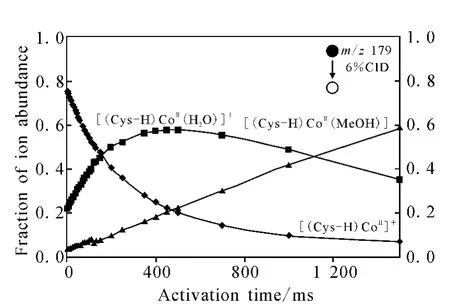
图7 在不同的CID激发时间下,配位化合物离子[(Cys-H)CoII]+及其捕获配体MeOH/H2O后的[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+三者之间丰度的变化Fig.7 Under the varied CID collision time to the target complex[(Cys-H)CoII]+ ,the fraction of ion abundance from[(Cys-H)CoII]+ ,[(Cys-H)CoII(H2O)]+and[(Cys-H)CoII(MeOH)]+
比较图5,6和7,当CID的激活时间超过200ms,3个配位化合物的热力学曲线非常吻合,[(Cys-H)CoII(MeOH)]+都保持着线形的持续增长,同时[(Cys-H)CoII(H2O)]+和[(Cys-H)CoII]+几乎平行的递减。
3 结论
在[(Cys-H)CoII(MeOH)]+,[(Cys-H)CoII(H2O)]+和[(Cys-H)CoII]+的 CID 试验中,研究了这3个配位化合物的相互转化过程。在CID的持续激发下,3个配位化合物离子会自发地按照钴离子配位化合物的热力学稳定性增加或减少,同时,受到离子阱中MeOH和H2O的相对含量及它们路易斯碱性的强弱差异的影响,气相系统最终达到 [(Cys-H)CoII(MeOH)]+含量最多,[(Cys-H)CoII(H2O)]+其次,[(Cys-H)CoII]+最少的平衡状态。
由此试验可推断,在[(Cys-H)CoII(MeOH)]+的 CID 中,[(Cys- H)CoII(MeOH)]+的数量会继续增加直到恢复到CID激发前的平衡。这也与[(Cys-H)CoII(MeOH)]+的热力学稳定性相一致:配位数为4的配位化合物[(Cys-H)CoII(MeOH)]+的内能在CID的激发下提高,而变为不稳定结构,通过释放配体MeOH减少内能。但生成的[(Cys-H)CoII]+具有不饱和配位键,从离子阱中捕获MeOH和H2O,回到结构更稳定且能量更低的[(Cys-H)CoII(MeOH)]+,并生成少量的[(Cys-H)CoII(H2O)]+。在[(Cys-H)CoII(MeOH)]+和[(Cys-H)CoII(H2O)]+之间的相互转化反应中,[(Cys-H)CoII]+起到了反应中间体的作用。
该实验表明,可以利用质谱分析仪的电喷雾(ESI)源产生气态配位化合物,利用离子阱的多级CID技术提高配位化合物的内能,从而破坏配位平衡,利用离子阱中I-M反应能力及其对离子的检测能力来研究气态中的金属配位化合物和配位平衡。
[1] HOGG A M,HAYNES R M,KEBARLE P.Ionsolvent molecule interactions studied in the gas phase.Heats and entropies of individual steps.NH4+·(n-1)NH3+NH3=NH4+·nNH3[J].J Am Chem Soc,1966,88(1):28-31.
[2] CASTLEMAN A W,TANG I N.Heavy metal ion clusters[J]. Nature,1971,234 (50):129-130.
[3] MCLUCKEY S A,CAMERON D,COOKS R G.Proton affinities from dissociations of protonbound dimers[J].J Am Chem Soc,1981,103(6):1 313-1 317.
[4] COOKS R G,PATRICK J S,KOTIAHO T,et al.Thermochemical determinations by the kinetic method[J].Mass Spectrom Rev,1994,13(4):287-339.
[5] COOKS R G,WONG P S H.Kinetic method of making thermochemical determinations:Advances and applications[J].Acc Chem Res,1998,31(7):379-386.
[6] CHENG X H,WU Z C,FENSELAU C.Collision energy-dependence of proton-bound dimer dissociation-entropy effects,proton affinities,and intramolecular hydrogen-bonding in protonated peptides[J].J Am Chem Soc,1993,115(11):4 844-4 848.
[7] CERDA B A,HOYAU S,OHANESSIAN G,et al.Na+binding to cyclic and linear dipeptides.Bond energies,entropies of Na+complexation,and attachment sites from the dissociation of Na+-bound heterodimers and ab Initio calculations[J].J Am Chem Soc,1998,120(10):2 437-2 448.
[8] WONG P S H,MA S G,COOKS R G.Ionization energy determination by the kinetic method[J].A-nal Chem,1996,68(23):4 254-4 256.
[9] BEAUCHAMP J L,FOSTER M S.Potential of ion cyclotron resonance spectroscopy for the study of the intrinsic properties and reactivity of transition metal complexes in the gas phase.Ion-molecule reactions of iron pentacarbonyl[J].J Am Chem Soc,1971,93(19):4 924-4 926.
[10] JAYAWEERA P,BLADES A T,IKONOMOU M G,et al.Production and study in the gas phase of multiply charged solvated or coordinated metal ions[J].J Am Chem Soc,1990,112(6):2 452-2 454.
[11] VACHET R W,HARTMAN J A R,CALLAHAN J H.Ion-molecule reactions in a quadrupole ion trap as a probe of the gas-phase structure of metal complexes[J].J Mass Spectrom,1998,33(12):1 209-1 225.
[12] VACHET R W,HARTMAN J R,GERTNER J W,et al.Investigation of metal complex coordination Structure using collision-induced dissociation and ion-molecule reactions in a quadrupole ion trap mass spectrometer[J].Int J Mass Spectrom,2001,204(1/2/3):101-112.
[13] UPPAL J S,STALEY R H.Relative binding energies of organic molecules to Mn+in the gas phase[J].J Am Chem Soc,1982,104(5):1 238-1 243.
[14] KAPPES M M,STALEY R H.Relative bond dissociation energies for two-ligand complexes of nickel(+)with organic molecules in the gas phase[J].J Am Chem Soc,1982,104(7):1 813-1 819.
[15] ANDERSEN U N,BOJESEN G.Gas-phase ion/molecule reactions of doubly charged clusters of Fe2+or Mn2+and methanol with ammonia[J].Int J Mass Spectrom ,1996,153(1):1-7.
[16] NIELSEN S B,BOJESEN G.Ligand and H/D exchange of Mn(H2O)62+and Cu(OH)(H2O)4+in the gas phase[J].Chem Commun,1998,(5):613-614.
[17] DENG H,KEBARLE P.Bond energies of copper ion-ligand L complexes CuL2+determined in the gas phase by ion-ligand exchange equilibria measurements[J].J Am Chem Soc,1998,120(12):2 925-2 931.
[18] ALVAREZ E J,VARTANIAN V H,BRODBELT J S.Dissociation of polyether-transition metal ion dimer complexes in a quadrupole ion trap[J].J Am Soc Mass Spectr,1997,8(6):620-629.
[19] LAVANANT H,HECQUET E,HOPPILLIARD Y.Complexes of L-histidine with Fe2+,Co2+,Ni2+,Cu2+,Zn2+studied by electrospray ionization mass spectrometry[J].Int J Mass Spectrom,1999,(185/186/187):11-23.
[20] COX H,AKIBO-BETTS G,WRIGHT R R,et al.Solvent coordination in gas-phase [Mn ·(H2O)n]2+and [Mn·(ROH)n]2+complexes:Theory and experiment[J].J Am Chem Soc,2003,125(1):233-242.
猜你喜欢
能源工程(2021年1期)2021-04-13
无机化学学报(2020年7期)2020-07-20
中国特种设备安全(2020年11期)2020-06-09
当代陕西(2019年6期)2019-04-17
科学之谜(2018年9期)2018-12-17
分析化学(2018年2期)2018-03-02
科技创新导报(2016年30期)2017-03-15
外语学刊(2014年3期)2014-12-03
郑州大学学报(理学版)(2013年2期)2013-03-11
中国科技信息(2012年11期)2012-10-26
