
蛋白质胰蛋白酶水解过程双位点漏切肽段的质谱鉴定
2013-05-28 07:23李水明何曼文邹永东
质谱学报 2013年1期
王 勇,李水明,何曼文,邹永东
(1.深圳大学生命科学学院,深圳市海洋生物资源与生态环境重点实验室,广东 深圳 518060;2.深圳大学生命科学学院,深圳市微生物基因工程重点实验室,广东 深圳 518060)
基于质谱鉴定蛋白分为自下而上(bottom up)和自上而下(top down)两种技术路线[1],前者是指将蛋白质酶解成肽段后进行质谱分析进而推测出整个蛋白质结构,后者则是直接分析完整蛋白。尽管top down方法给出信息更为直接可靠并发展很快,但由于对质谱仪的分辨率、样品纯度和离子断裂方式等要求很高,目前大部分蛋白质组学研究都采用bottom up技术或者将二者结合[2-6],除非基于特殊目的[7],使用的多为胰蛋白酶,它能够特异性地酶解赖氨酸(Lys,K)和精氨酸(Arg,R)残基的羧基端。
使用一定的算法或软件将质谱数据与蛋白质数据库匹配是蛋白质组学的核心[8],算法编程遇到的问题之一是非特异性酶切和漏切[9]。尽管Olsen等基于高分辨率质谱的数据认为胰蛋白酶的酶切特异性很高,之前文献报道的大部分胰酶非特异性酶切或者半酶切很可能是因为自动搜库导致的伪结果[10],但胰酶漏切现象在蛋白质的鉴定中并不罕见。早期研究者利用104个蛋白质组成的数据集证明当赖氨酸和精氨酸彼此或自身相邻、或它们各自与天冬氨酸或谷氨酸相邻、或赖氨酸/精氨酸后面(羧基端)为脯氨酸时将会发生漏切[11]。目前,只有关于脯氨酸漏切原则被广泛接受编入许多搜库算法,但也有工作表明这一原则在很多情况下会被违反[12]。总体上,影响胰酶水解特异性的因素有很多,例如实验条件和裂解位点附近的残基等,但文献报道中关于胰酶漏切位点的研究大多基于生物信息学和蛋白质组学方法展开[13-14],针对单一蛋白研究胰酶漏切的工作较少。Mascot软件的应用指南中指出漏切位点数可以为1或2,但未见更详细的说明。本研究利用基质辅助激光解吸电离-串联飞行时间质谱(MALDI-TOF/TOF)分析细胞色素C和大豆脱落胁迫成熟蛋白,为更好地了解胰酶漏切现象提供实验依据。
1 实验部分
1.1 仪器与试剂
4800MALDI TOF/TOFTMAnalyzer基质辅助激光解吸电离-串联飞行时间质谱仪(MALDI-TOF/TOF):美 国 Applied Biosystems/MDX SCIEX公司产品;超纯水处理系统(Milli-Q):美国 Millipore公司产品。
胰蛋白酶(质谱分析级):Promega公司产品;α-腈基-4-羟基肉桂酸:美国Sigma公司产品,使用前重结晶。蛋白分子量标记物#SM0431(包括溶菌酶等7种标准蛋白质):美国Thermo Fisher Scientific公司产品。
1.2 胶内胰酶水解条件
蛋白分子量标记物经一维电泳分离后的目标条带用移液枪枪头切下,小心地放入PCR管,用100μL高纯水振荡洗涤10min,100μL 25 mmol/L NH4HCO3室温振荡平衡20min;100 μL 25mmol/L NH4HCO3/50%乙腈室温振荡30min;100μL 25mmol/L NH4HCO3室温振荡平衡20min;加入100μL 100%乙腈于胶粒中,静置10min,重复1次;加入5μL 20mg/L质谱测序级胰蛋白酶,4℃放置30min;加入5 μL 40mmol/L NH4HCO3/10%ACN,37 ℃水浴8h。
1.3 质谱分析条件
基质制备:将6g/Lα-腈基-4-羟基肉桂酸和2g/L柠檬酸氢二胺溶于10mL 50%乙腈(含0.1%三氟乙酸),用V(基质溶液)∶V(蛋白酶解液)=1∶1的溶液混合后点于不锈钢靶版,自然干燥结晶后待测。采用正离子反射模式,一级质谱每张谱图累加800次,二级质谱累加1 200次,碰撞诱导解离条件为1kV碰撞能加空气碰撞(1kV,gas on)。
1.4 数据分析条件
数据库搜索条件:串联质谱数据搜库,信噪比设为5,一级和二级质谱质量误差均设为±0.3Da,Mascot软件分析,搜索数据库 NCBI2012年8月19日发布,甲硫氨酸氧化(Oxidation)和半胱氨酸烷基化(Carbamidomethyl)作为可变修饰,胰酶漏切位点数1或2。
2 结果与讨论
2.1 细胞色素C的胰蛋白酶的双位点漏切
细胞色素C胰蛋白酶水解液的一级质谱示于图1,一级质谱中各[M+H]+离子做串联质谱分析,以串联质谱数据搜库的Mascot得分是657分,证明该蛋白确为细胞色素C。但是,图1中m/z1 478.96和m/z2 209.33离子未被数据库匹配,为确定结构,对它们的二级谱图进行进一步分析。
图2和图3的共同点是都在低质量区观察到了较高丰度的m/z84.09离子,在图3中m/z129离子的相对强度也较高,而这两个离子是赖氨酸残基的亚胺相关离子[15],所以猜测它们有可能是细胞色素C不完全酶解的产物。因为应用漏切数为1的常规搜库设置条件时没有匹配,将漏切位点数目设为2后重新搜库,m/z1 478.96被 鉴 定 为 KTEREDLIAYLK,m/z2 209.33离 子 的 序 列 为 GITWKEETLMEYLENPKK,它们都含有两个胰蛋白酶酶切漏点,Mascot分数分别为82和61,鉴定结果可靠。值得注意的是,肽段TEREDLIAYLK和GITWKEETLMEYLENPK在常规条件下已被检出,因此二次搜库的结果只相当于多鉴定了两个赖氨酸残基,鉴定覆盖率从72%提高到74%(细胞色素C由101个氨基酸组成),但是,鉴定的Mascot分数从657增至779。

图1 细胞色素C胰蛋白酶水解液的MALDI-TOF质谱图Fig.1 MALDI-TOF spectrum of tryptic digest of cytochrome C
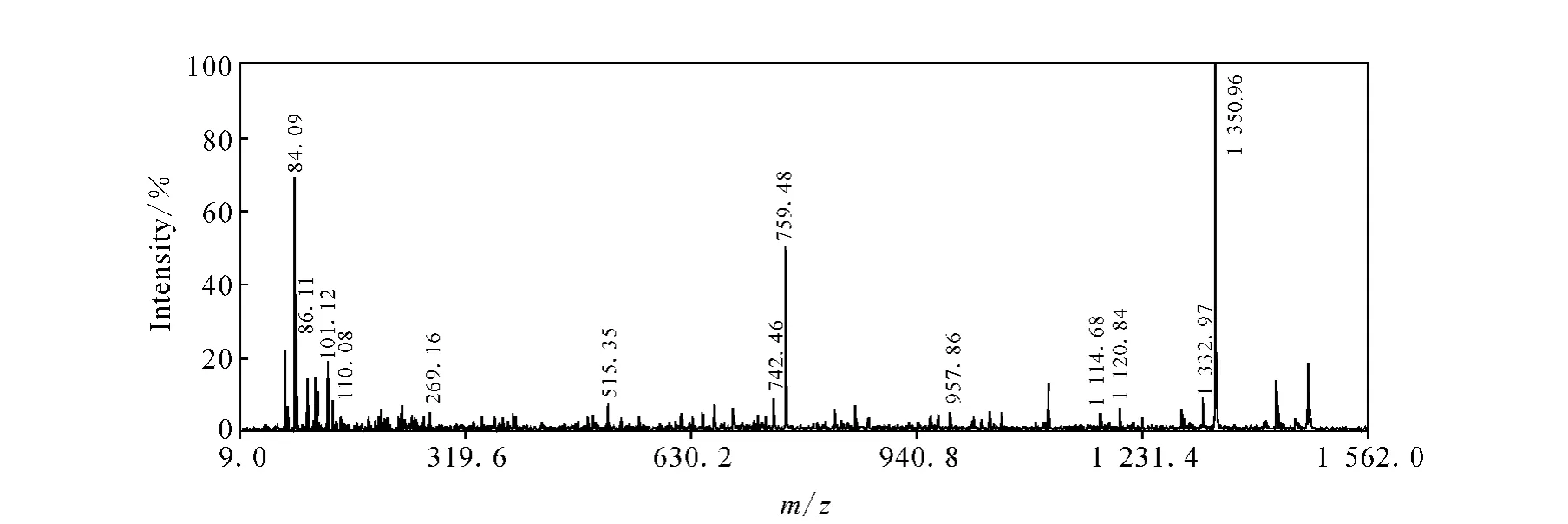
图2 肽段[M+H]+ m/z 1 478.96离子的串联飞行时间质谱图Fig.2 MALDI-TOF/TOF spectrum of peptide[M+H]+ m/z 1 478.96
2.2 大豆脱落胁迫成熟蛋白的胰蛋白酶的双位点漏切
类似地,将胰酶漏切位点数设为2后,在另外的一个实际样品分析中也检测到了多个双酶解漏切肽段,示于图4。在所有其他搜库条件不变的情况下将胰酶漏切位点个数由1变为2后,鉴定蛋白的 Mascot得分由原来的1 008变为1 558,覆盖率由46%增加至48%。Mascot分值显著增加而覆盖率增加较小,这与样品蛋白的序列特性和软件的打分算法有关,前者说明鉴定蛋白的确定性显著提高,而覆盖率增加较小则说明有重合肽段。

图3 肽段[M+H]+ m/z 2 209.33离子的串联飞行时间质谱图Fig.3 MALDI-TOF/TOF spectrum of peptide[M+H]+ m/z 2 209.33
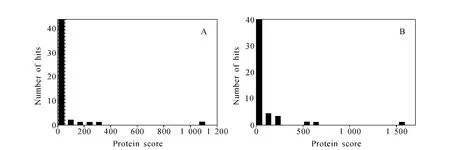
图4 大豆脱落胁迫成熟蛋白串联质谱数据的Mascot搜库结果Fig.4 The database searching result of abscisic stress ripening-like protein by Mascot
大豆脱落胁迫成熟蛋白的大多数水解肽段都有漏切现象,实验中将漏切数设为0,则只匹配3条肽段,搜库分数为379,其搜库鉴定结果列于表1。值得注意的是,发生双位点漏切的4个肽段都有相应的单位点漏切肽段,分别是EQDEEAHGKK/EAKEQDEEAHGKK,AEKDPEHAHR/HKAEKDPEHAHR,TSGGGYDDEVDYKK/TSGGGYDDEVDYKKEEK和 HKIEEEVAAAAAVGSGGFAFHEHHEK/HKIEEEVAAAAAVGSGGFAFHEHHEKK。对漏切位点进行分析,发现漏切现象主要发生在赖氨酸残基,很多是由于存在两个连续的赖氨酸或者赖氨酸与天冬氨酸或谷氨酸相邻。胰酶水解位点的漏切还可能与酶解时间有关,但是本实验水解时间均为8h,并且细胞色素C和大豆脱落胁迫成熟蛋白不是同一批处理的样品,在它们各自的同批次蛋白样品中也未观察到漏切肽段,因此推测双位点漏切与蛋白的结构有关。
在该例中不仅证实了文献[11]中提及的大部分漏切现象,还观察到了HKA和HKI结构引起的漏切(表1),并呈现相对定量的关系。如图5所示,肽段HKAEKDPEHAHR[M+H]+峰(m/z1 454.78)被作为基峰检测,而对应肽段AEKDPEHAHR(m/z1 189.60)的相对强度为28.6%。此外,肽段HKIEEEVAAAAAVGSGGFAFHEHHEK的[M+H]+峰(m/z2 758)的相对强度远高于HKIEEEVAAAAAVGSGGFAFHEHHEKK (m/z2 886)和IEEEVAAAAA VGSGGFAFHEHHEK(m/z2 493),该结果提示,即使经过长时间的酶解过程,含有HKA和HKI结构的肽段仍很稳定,具体原因尚需进一步研究。此外,将漏切位点数设为2搜索没有发现额外的假阳性。
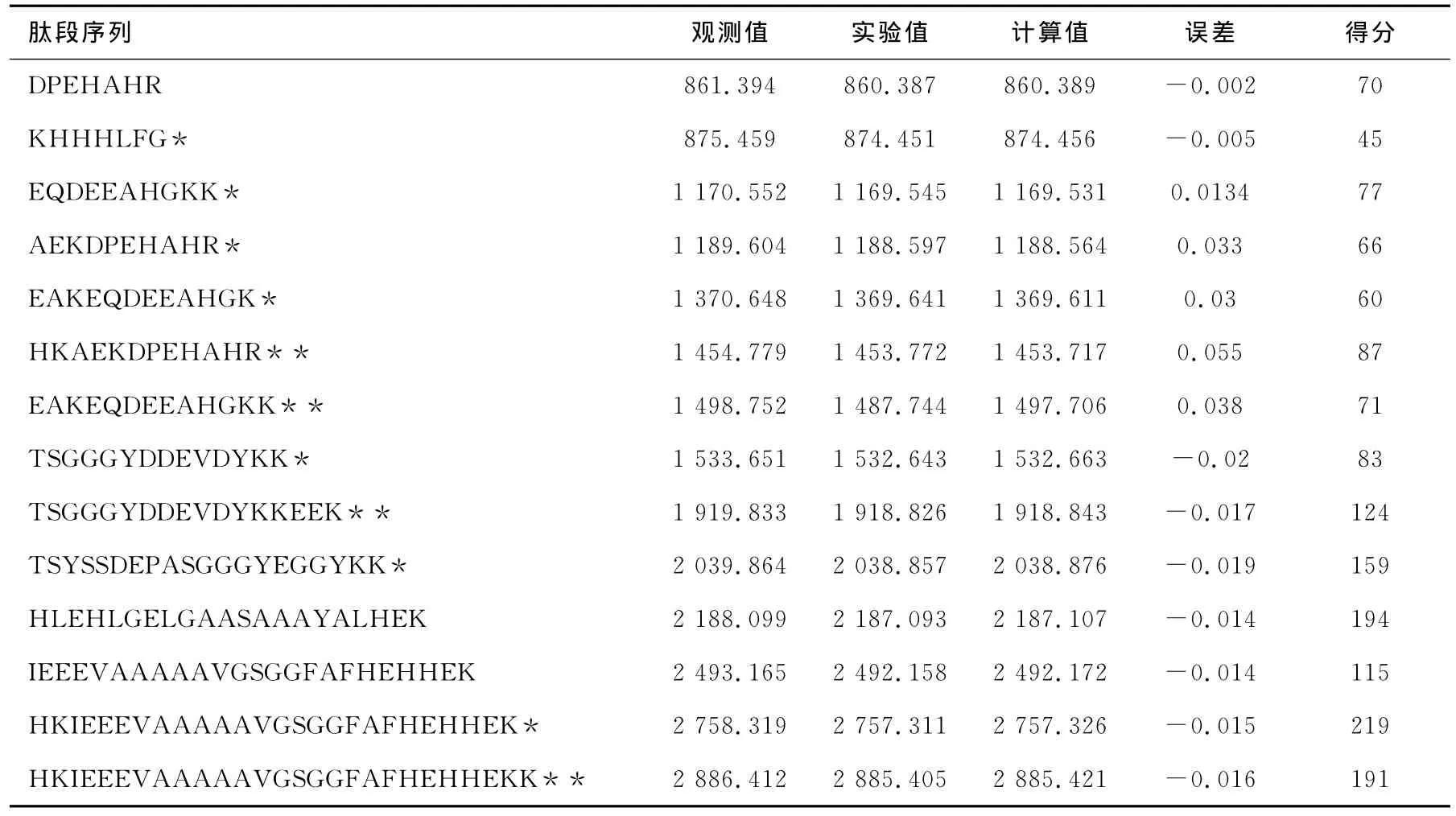
表1 大豆脱落胁迫成熟蛋白的搜库鉴定结果Table 1 Peptides of abscisic stress ripening-like protein[Glycine max]identified by Mascot

图5 大豆脱落胁迫成熟蛋白胰蛋白酶水解液的MALDI-TOF质谱图Fig.5 MALDI-TOF spectrum of tryptic digest of abscisic stress ripening-like protein
3 结论
在常规实验条件下的细胞色素C和大豆脱落胁迫成熟蛋白体系中,发现和确认了胰蛋白酶的双位点漏切现象,增加漏切位点数目搜索后覆盖率无明显提高,但显著增加了搜库分数及鉴定结果的可信度。发现胰酶漏切现象与蛋白质结构关系密切,具有一定的规律性。本工作表明对于某些序列特殊的蛋白质,如果漏切位点设置不当可能导致数据利用率降低。
[1] BOGDANOV B,SMITH R D.Proteomics by FTI-CR mass spectrometry:Top down and bottom up[J]. Mass Spectrom Review,2005,24(2):168-200.
[2] CHI H,SUN R X,YANG B,et al.pNovo:De novo peptide sequencing and identification using HCD spectra[J].Journal of Proteome Research,2010,9(5):2 713-2 724.
[3] MEDZIHRADSZKY K F,CAMPBELL J M,BALDWIN M A,et al.The characteristics of peptide collision-induced dissociation using a high-performance MALDI-TOF/TOF tandem mass spectrometer[J].Analtical Chemistry,2000,72(3):552-558.
[4] MA M M,CHEN R B,GE Y,et al.Combining bottom-up and top-down mass spectrometric strategies for de novo sequencing of the Crustacean hyperglycemic hormone from cancer borealis[J].Analtical Chemistry,2009,81(1):240-247.
[5] MIYASHITA M,HANAI Y,AWANE H,et al.Improving peptide fragmentation by N-terminal derivatization with high proton affinity[J].Rapid Communication Mass Spectrometry,2011,25(9):1 130-1 140.
[6] HUANG L,BALDWIN M A,MALTBY D A,et al.The identification of protein-protein interactions of the nuclear pore complex of saccharomyces cerevisiae using high throughput matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry[J]. Molecular Cell Proteomics,2002,(1):434-450.
[7] BOERSEMA P J,TAOUATAS N,MAARTEN A F,et al.Straightforward and de novo peptide sequencing by MALDI-MS/MS using a Lys-N metalloendopeptidase[J].Molecular Cell Proteomics,2009,(8):650-660.
[8] MARCOTTE E M.How do shotgun proteomics algorithms identify proteins[J].Nature Biotechnology,2007,25(7):755-757.
[9] LUBEC G,AFJEHI-SADAT L.Limitations and pitfalls in protein identification by mass spectrom-etry[J].Chem Rev,2007,107:3 568-3 584.
[10] OLSEN J V,ONGS E,MANN M.Trypsin cleaves exclusively C-terminal to arginine and lysine residues[J].Molecular Cell Proteomics,2004,(3):608-614.
[11] THIEDE B,LAMER S,MATTOW J,et al.Analysis of missed cleavage sites,tryptophan oxidation and N-terminal pyroglutamylation after in-gel tryptic digestion[J].Rapid Communication Mass Spectrometry,2000,14(6):496-502.
[12] RODRIGUEZ J,GUPTA N,RICHARD D,et al.Does trypsin cut before proline[J].J Proteome Research,2008,7(1):300-305.
[13] SIEPHENJ A,KEEVIL E J,KNIGHT D,et al.Prediction of missed cleavage sites in tryptic peptides aids protein identification in proteomics[J].J Proteome Research,2007,6(1):399-408.
[14] 刘 辉,张纪阳,孙汉昌,等.基于马尔科夫链的胰蛋白酶肽段蛋白酶切位点概率预测[J].生物物理学报,2011,27(6):528-536.
[15] FALICK A M,HINES W M,MEDZIHRADSZKY K F,et al.Low-mass ions produced from peptides by high-energy collision-induced dissociation in tandem mass spectrometry[J].Journal of the American Society for Mass spectrometry,1993,4(11):882-893.
猜你喜欢
北方药学(2022年3期)2022-09-06
新世纪智能(英语备考)(2018年11期)2018-12-29
现代装饰(2018年1期)2018-05-22
中西医结合心血管病电子杂志(2016年36期)2017-07-05
制造技术与机床(2017年3期)2017-06-23
科学与财富(2017年11期)2017-05-25
探测与控制学报(2015年4期)2015-12-15
中国畜牧业(2015年6期)2015-12-07
中国兽药杂志(2015年12期)2015-01-25
中国畜牧业(2014年5期)2014-12-20
