
一种基于iSRM策略的蛋白质组质谱数据分析工具
2013-05-28 07:23卫军营徐长明张纪阳朱云平钱小红张养军谢红卫
质谱学报 2013年1期
张 伟,卫军营,贾 婷,徐长明,张纪阳,朱云平,钱小红,张养军,谢红卫
(1.国防科学技术大学,机电工程与自动化学院自动控制系,湖南 长沙 410073;2.军事医学科学院放射与辐射医学研究所,北京蛋白质组研究中心,蛋白质组学国家重点实验室,北京 102206;3.山东省中医药大学,第二附属医院生殖医学中心,山东 济南 250001)
蛋白质组学旨在系统研究细胞、组织或生物体全套蛋白质的组成和功能,以及在不同生理病理条件下量的变化。一般来说,生物样本可包含成千甚至上万个不同的蛋白质,并且这些蛋白质的相对含量可能超过5个数量级,这给蛋白质组学研究带来了不小的技术挑战。目前,质谱分析技术是实现大规模、高通量蛋白质定性和定量分析的主要方法[1]。在基于质谱技术的蛋白质组学研究中,“鸟枪法”(shotgun approach)是常用的实验策略[2-3]。尽管已经有很多成功的应用,但“鸟枪法”的技术局限不容忽视。面对复杂样本,“鸟枪法”肽段鉴定的重现性较差,对低丰度肽段的监测灵敏性较差,并且其定量水平还达不到基因芯片技术在转录组定量分析的水平[4]。
选择反应监测技术(selected reaction monitoring,SRM)在一定程度上弥补了“鸟枪法”的上述缺陷[4-6]。SRM 是一种目标蛋白质组学(target proteomics)研究策略,实验通常在三重四极杆串联质谱仪(triple quadrupole system,QQQ)[7-8]上进行。首先,预先设定目标蛋白质的特定母离子和子离子对;然后,根据选择的离子对,分别在QQQ的第一级(Q1)中选择肽段母离子,在第二级(Q2)中碎裂母离子,在第三级(Q3)中监测对应的子离子;最后依据子离子的峰面积,实现对目标蛋白质的定量分析。通过在母离子和子离子两个水平排除干扰,增加了监测的灵敏性、重现性以及准确性,可以用来进一步验证候选的差异蛋白质。另一方面,肽段通常需要监测到8~10个子离子才能充分证明其存在[9],即在常规的SRM策略中,对于每个肽段一般需要监测8~10个“母离子-子离子”对。在Q2中碎裂肽段母离子是非常耗时的,那么在实验时间一定的情况下,可监测的目标肽段比较少。为了弥补上述缺陷,2011年,Kiyonami等提出了智能选择反应监测(intelligent selected reaction monitoring,iSRM)的 新 型 实 验 策略[10]。iSRM策略把每个肽段的所有“母离子-子离子”对分成两个部分:一是主要的(primary)定量子离子(一般设为2),另一是次要的(secondary)定性子离子。其中在整个分析时间内,一直对定量子离子进行监测,当定量子离子的信号强度同时达到设定的阈值,触发监测定性子离子,用来对定量肽段的可靠性进行验证。iSRM策略减少了每个肽段监测定性子离子的次数,从而增加了可监测目标肽段的数目。
目前,处理基于SRM策略质谱数据的软件很多,如 mProphet[11]、Skyline[12]、ATAQS[13]、TIQAM[14]和 MRMer[15]等,但这些软件大都是基于常规的SRM策略编写的数据处理软件,而针对iSRM策略的数据处理工具却很少见。实验人员一般利用Thermo Scientific公司的Pin-Point和Xcalibur软件人工获得每个肽段的定量信息,这非常耗费时间和精力。本工作利用MATLAB编程语言设计了针对iSRM策略的蛋白质组质谱数据的自动处理软件——iSQuant。下面首先详细介绍iSQuant的数据处理算法流程;然后利用3组iSRM质谱数据验证iSQuant的可重复性能和线性性能。
1 方法与材料
1.1 iSRM实验数据
本实验用于评估iSQuant的实验数据来自北京蛋白质组研究中心[16],其中包括了小鼠(mouse)的重复实验数据、酵母(yeast)的重复实验数据以及牛血清白蛋白(bovine serum albu-min,BSA)的标准实验数据。两组重复实验数据都包含3次技术重复实验;标准数据集包含5个批次的上样量(分别为1、10、100、500和1 000fmol),其中每个批次有2次技术重复实验。
1.1.1 样本制备 小鼠样本的制备来自Song等[17]的样本制备流程。
对于酵母的样本制备,首先将200mg酵母悬浮于1mL含50mmol/L NH4HCO3的裂解液中,在冰浴上超声裂解(每超声0.1s,暂停2s,重复100次)。室温静置30min后,将裂解液在40 000r/min下离心60min,再将上清液于40 000r/min下离心60min,用Bradford法测定蛋白浓度为1.46g/L。在蛋白提取物(100 μg,68.4μL)中加入 25μL 溶于 50mmol/L NH4HCO3的8mol/L尿素溶液进行蛋白变性后,加入2.78μL 100mmol/L的DTT,在37℃水浴反应4h,再加入1.46μL 1mol/L的IAA在室温暗处反应1h。用50mmol/L NH4HCO3溶液稀释,使脲的最终浓度小于1mol/L之后,加入V(胰酶)∶V(底物)=1∶50的胰酶在37℃水浴中酶切过夜。酶解液浓缩后重溶于2%乙腈(含0.1%甲酸)溶液。
对于 BSA 的样本制备,用50mmol/L NH4HCO3溶液对BSA溶解后,加入DTT(终浓度10mmol/L)于95℃水浴孵育10min。再加入IAA(终浓度50mmol/L)于室温暗处反应1h。最后加入V(胰酶)∶V(底物)=1∶50的胰酶在37℃水浴中酶切过夜。
1.1.2 色谱-质谱条件 Eksigent 2D 液相系统:Eksigent公司产品;在线脱盐trap柱(u-Precolumn Cartridge,0.3mm×5mm):戴安公司产品;自制毛细管直喷柱(75μm×150mm,填料 GEAgel,SP-300-ODS-AP,5μm,金欧亚公司产品)。线性梯度为95%A相(2%乙腈、0.1%甲酸溶液)-50%B相(80%乙腈、0.1%甲酸溶液),梯度时间1h,流速300nL/min。
触发采集二级质谱(quantitation-enhanced data-dependent MS/MS)和iSRM 采集方法:正离子模式,喷射电压2.1kV,毛细管温度200℃,Q1和Q3分辨率设置为0.7(FWHM),Q2碰撞氩气压力19.03Pa,触发阈值设定为100,碰撞能量(CE)按如下公式计算:CE=(0.034×母离子的质荷比)+2.314。iSRM方法循环时间(cycle time)设置为2s,触发iSRM 采集时间设置为0.1s。
1.2 iSQuant的数据处理流程
iSQuant的数据处理流程示于图1。在实验阶段,蛋白质样本酶切成肽段混合物,并且根据预测工具(如STEPP、ESP Predictor等)或者SRM 数据库(如SRMAtlas、MRMaid-DB等)给出目标蛋白质的Transition对列表;然后,肽段混合物进入QQQ质谱仪,根据Transition列表在质谱仪中设定Transition信息,并且设定iSRM模式可获取RAW格式的iSRM数据文件。在数据处理阶段,首先将RAW格式的文件转换成_mrml.xml格式文件;然后,读取_mrml.xml文件的信息,通过提取每条肽段的定量子离子的定量信息,估计该肽段的相对丰度。在提取定量子离子的定量信息之前,需要区分目标肽段的定性子离子和定量子离子,若目标肽段的定性子离子未被触发(即没有定性子离子),那么认为该肽段监测到的定量子离子不可靠;反之,对定量子离子的离子流色谱峰(extracted ion chromatogram,XIC)进行峰监测并提取其定量信息。上述数据处理流程都是在MATLAB中实现的。
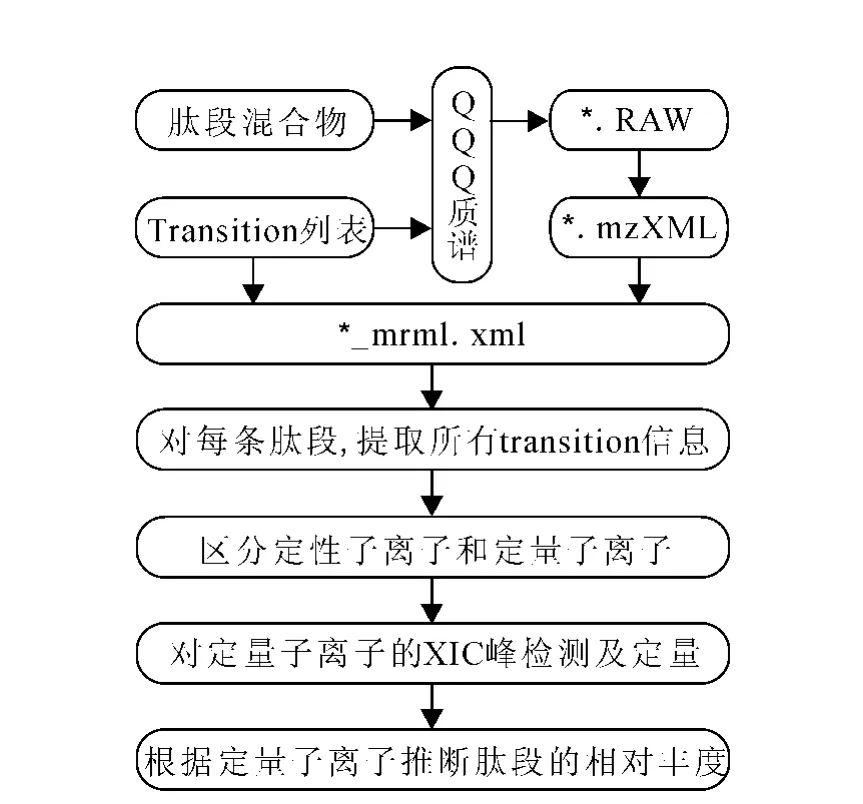
图1 iSQuant的数据处理流程Fig.1 Data processing workflow of iSQuant
1.3 数据格式转换
为了将RAW格式的iSRM数据文件转换成_mrml.xml格式数据文件,首先,应该将RAW格式文件转为mzXML格式文件,推荐使用ReAdW.exe数据转换工具,下载网址为http://tools.proteomecenter.org/wiki/index.php?title=Software:ReAdW;在cmd中输入命令行:
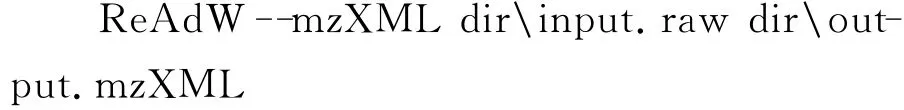
其中,dir表示存放RAW和mzXML文件的文件路径名。然后,使用mProphet软件[11]中的数据转换工具mMap2.pl将mzXML和transition列表整合成_mrml.xml格式数据文件;在命令行中输入:
mMap2.pl-xmls dir\input.mzXML -mach TSQ -strict-trans dir\TransitionList.txt
其中,dir表示存放RAW和mzXML文件的文件路径名,产生的_mrml.xml文件也将自动存放在dir文件路径中。值得注意的是,_mrml.xml文件不仅包含了mzXML文件的质谱数据信息,而且还包含了transition的“蛋白质-肽段母离子-子离子”对应关系。
1.4 肽段的相对丰度估算
针对_mrml.xml格式数据文件,BSA肽段AEFVEVTK的定量子离子的定量过程示于图2。
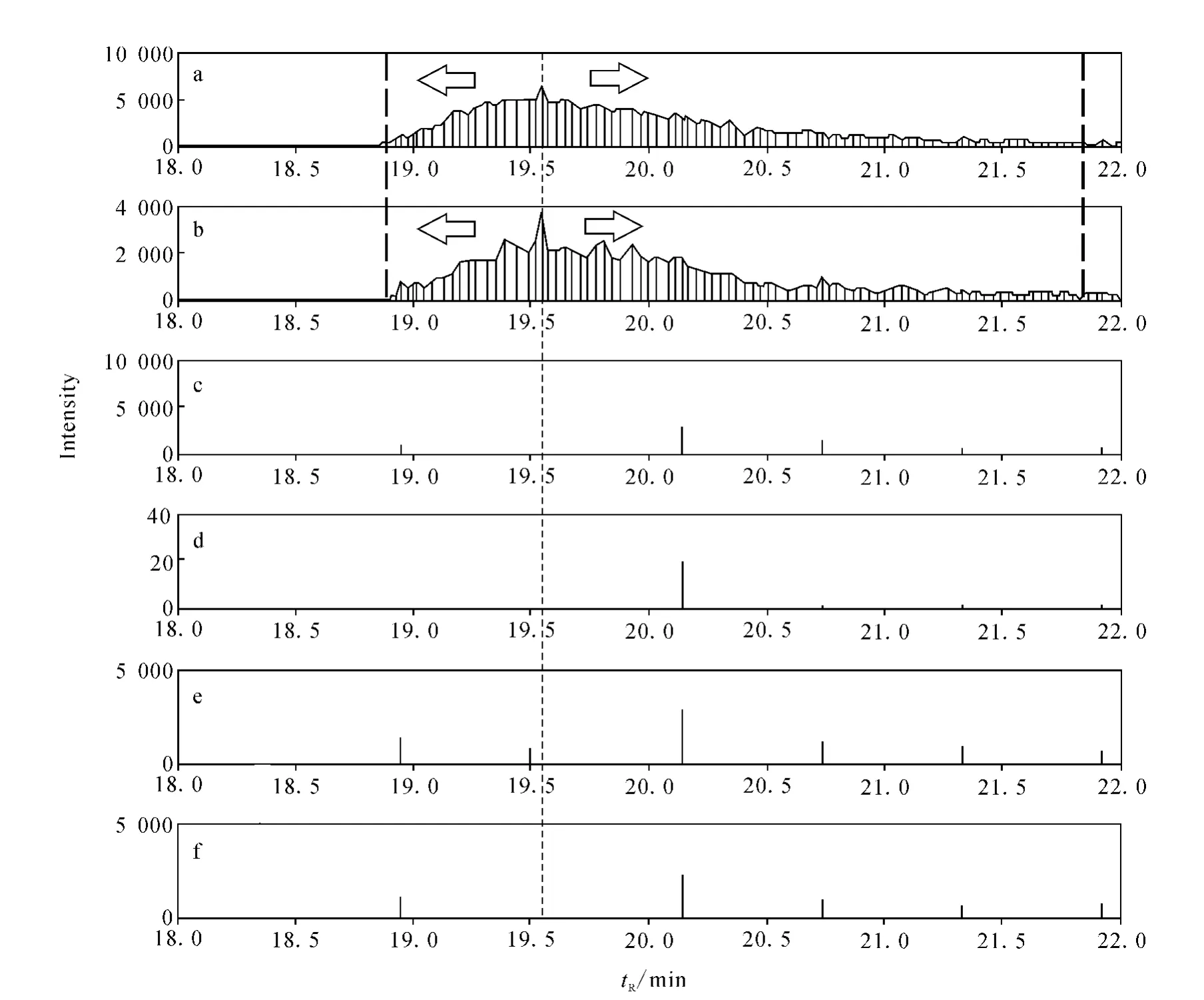
图2 BSA肽段AEFVEVTK定量离子的定量过程Fig.2 Quantitative process of BSA peptide AEFVEVTK
肽段的相对丰度估算的算法步骤可描述如下。
1)对每条肽段,提取其对应的所有transition信息,包括肽段母离子与子离子的对应关系、子离子在不同色谱流出时间处的信号强度信息。
2)从上述子离子中区分出定量子离子和定性子离子。若没有定性子离子被触发,则表明监测到的定量子离子不可靠。在iSRM策略中,通过设定延迟时间和动态排除时间,当两个定量子离子的信号同时达到仪器设定的阈值时才触发监测定性子离子的信号强度。一般情况下,肽段在一次实验中其定性子离子可被触发4~6次左右,而定量子离子在每个采样循环中都需要监测。因此可以根据子离子在色谱流出时间轴上的数据点数来区分定量子离子(图2a和2b)和定性子离子(图2c~2f)。
3)根据定性子离子来确定定量子离子大致的色谱保留时间(retention time,RT)。如步骤2中的描述,仪器需要设定延迟时间来触发定性子离子,第一次触发监测定性子离子的时间等于第一次达到信号强度阈值的时间加上延迟时间。这样做的好处是尽量使触发时间发生在定量子离子XIC的最高峰附近,那么可以根据定性子离子的触发时间来确定定量子离子大致的色谱保留时间。具体做法是,选择定性子离子中信号强度最强的触发时间作为定量子离子的色谱保留时间(图2中细虚线标识的位置)。
4)对定量子离子进行XIC峰检测,并估计其定量值。具体做法如下:首先对定量子离子的信号强度进行Savitzky-Golay去噪处理;其次求取定量子离子在色谱流出时间轴上的所有局部最大值和局部最小值;然后以其RT为起点,在色谱流出时间轴上分别向左和向右查找XIC峰的左右端点,当遇到的局部最小值小于最大值的0.05倍,或者遇到的局部最大值大于前一个局部最大值时,则找到了端点(图2a和2b中的粗虚线);最后利用梯形法求取XIC峰的峰面积,用峰面积值来估计定量子离子的定量值。
5)根据定量子离子的定量值推算肽段的相对丰度。通常情况下,每条肽段都有两个定量子离子,可以使用这两个定量子离子的定量值的加和来估计其相对丰度。
2 结果与讨论
可重复性能(reproducibility)和线性性能(linearity)通常被用来考察定量方法的定量性能[18-19]。为了考察iSQuant的定量性能,本研究利用小鼠的3次重复实验数据和酵母的3次重复实验数据来评估iSQuant的可重复性能,以及利用BSA标准实验数据来评估iSQuant的线性性能。
2.1 重复性能评估
对于小鼠的3次重复实验,选择了小鼠蛋白质的122条酶切肽段,其中每条肽段选取2个定量子离子和4~6个定性子离子。利用iSQuant提取定量信息,共有106条肽段在3次重复实验中都触发了定性离子的产生,其定量信息可视为可信的肽段定量估计值。此外,还利用了Xcalibur看图软件,采用人工验证的方法获得了106条肽段在3次重复实验中的定量估计值。人工验证的定量结果将作为参考标准,来衡量iSQuant的定量结果。为了考察可重复性能,首先求取每个肽段在3次重复实验中定量估计值的平均值,然后分别计算肽段在每次实验的定量估计值与平均值的线性相关系数。线性相关系数越接近于1,说明定量方法的可重复性能越好[20]。iSQuant定量结果在3次重复实验与平均值之间的线性相关展示图示于图3a~3c;人工验证的定量结果的线性相关展示图示于图3d~3f。由图3不难看出,iSQuant的3个线性相关系数都为0.97,而人工验证的相关系数则都为0.99。尽管iSQuant的相关系数略低于人工验证结果,但是iSQuant方法仍具有很高的可重复性能。比较iSQuant方法与人工验证方法的差异,虽然两种方法的重复性能都随丰度的降低而变差,但是iSQuant方法的这种趋势更加明显,这也是iSQuant的相关系数略低于人工验证方法的主要原因。这也说明,iSQuant的性能还有部分提升的空间,应该寻求一种更加灵敏的挑峰准则,缩小软件在低丰度肽段上带来的误差。
对于酵母的3次重复实验,选择了128个蛋白质中的202个目标肽段,其中每个肽段选取2个定量子离子和6个定性子离子,共包含1 608个母离子对。由于这128个蛋白质包含了许多低丰度的蛋白质[16],因此可能存在多条肽段的定性子离子不被触发。利用iSQuant提取定量信息,可得到69个可信的肽段定量估计值。为了衡量iSQuant的可重复性能,在数据归一化和取log10对数转换后,分别计算69个肽段在3次重复实验中定量估计值的变异系数(coefficient of variation,CV),CV 值分布与定量估计值均值之间的散点分布图示于图4。数据归一化采用了Valot等[21]提到的方法,即首先选择任意一个实验为参考实验(reference run);然后对每个非参考实验,计算这次实验与参考实验对应肽段的定量估计值的比值;最后通过除以上述比值的中值求得归一化以后的定量值。由图4可以得到,CV的均值低达1.27%,且CV值分布限制在一个狭小的范围内(0~2.5%)。极小的CV值分布不难说明,iSRM策略以及iSQuant工具具有很好的可重复性能。
2.2 线性性能评估
BSA的标准实验选择了BSA蛋白质的18条酶切肽段,其中每条肽段选取2个定量子离子和4~6个定性子离子。利用iSQuant提取定量信息,共有16条肽段在所有实验中都触发了定性离子的产生,其定量信息可视为可信的肽段定量信息。由于每个BSA实验数据的蛋白质上样量都已知(包含 1、10、100、500和 1 000fmol 5个批次的上样量,并且每个批次有2次技术重复实验),因此将已知的上样量为参考标准,来衡量iSQuant的定量结果。

图3 iSQuant在小鼠数据上的可重复性能评估Fig.3 Reproducibility estimation of iSQuant on mouse dataset

图4 iSQuant在酵母数据上的可重复性能评估Fig.4 Reproducibility estimation of iSQuant on yeast dataset
为了考察线性性能,首先对每个上样量,把每个肽段在两次重复实验中丰度的平均值作为肽段在这个上样量的丰度估计值;然后分别计算每个肽段的丰度估计值(log10)与实际上样量(log10)之间的相关系数。如果相关系数越接近于1,就说明定量方法的线性性能越好。BSA的16条肽段在5个上样量中丰度估计值(log10)以及相应的相关系数列于表1。不难得出,16条肽段的相关系数都大于0.92,所有相关系数的均值和方差分别为0.98和0.02,这表明iSQuant方法具有很好的线性性能。此外,还考察了iSQuant在蛋白质水平的线性性能,采用常规的蛋白质推算方法来计算BSA在每个上样量的丰度估计值(即通过计算16条肽段的丰度值之和获得)。BSA的丰度估计值与其实际的上样量之间的线性关系示于图5,其中相关系数高达0.99,同样表明了iSQuant优越的线性性能。

表1 BSA肽段在不同上样量丰度估计值及相关系数Table 1 Abundance estimation and correlation coefficient of BSA peptide among different loading amount

图5 BSA的丰度估计值与上样量的线性关系Fig.5 Linear correlation between BSA abundance estimation and loading amount
3 结论
针对新型的质谱选择反应监测(SRM)策略——iSRM策略产生的质谱数据,开发了一种肽段定量信息提取工具——iSQuant。该工具采用MATLAB脚本语言编写,简便易读。分别选用了小鼠的3次重复实验数据、酵母的3次重复实验数据和BSA的标准实验数据对iSQuant的可重复性能和线性性能进行了评估。结果表明,iSQuant的定量结果具有优越的重复性能和线性性能,基本达到了人工验证方法的性能要求。
[1] AEBERSOLD R,MANN M.Mass spectrometrybased proteomics[J].Nature,2003,422(6 928):198-207.
[2] YATES J R,RUSE C I,Nakorchevsky aproteomics by mass spectrometry:Approaches,advances,and applications[J].Annu Rev Biomed Eng,2009,11(1):49-79.
[3] 张 伟,张纪阳,刘 辉,等.蛋白质质谱分析的无标记定量算法研究进展[J].生物化学与生物物理进展,2011,38(6):506-518.
[4] LANGE V,PICOTTI P,DOMON B,et al.Selected reaction monitoring for quantitative proteomics:A tutorial[J].Mol Syst Biol,2008,4(10):222-235.
[5] PICOTTI P,AEBERSOLD R.Selected reaction monitoring-based proteomics:Workflows,potential,pitfalls and future directions[J].Nat Methods,2012,9(6):555-566.
[6] BRÖNSTRUP M.Absolute quantification strategies in proteomics based on mass spectrometry[J].Expert Rev Proteomics,2004,1(4):503-512.
[7] YOST R A,ENKE C G.Triple quadrupole mass spectrometry for direct mixture analysis and struc-tural elucidation[J].Anal Chem,1979,51(12):1 251-1 264.
[8] YOST R A,ENKE C G.Selected ion fragmentation with a tandem quadrupole mass spectrometer[J].J Am Chem Soc,1978,100(7):2 274-2 275.
[9] LIN D,TABB D L,YATES J R.Large-scale protein identification using mass spectrometry [J].Biochim Biophys Acta,2003,1 646(1/2):1-10.
[10] KIYONAMI R,SCHOEN A,PRAKASH A,et al.Increased selectivity,analytical precision,and throughput in targeted proteomics[J].Mol Cell Proteomics,2011,10(2):M110002931.
[11] REITER L,RINNER O,PICOTTI P,et al.mP-rophet:Automated data processing and statistical validation for large-scale SRM experiments[J].Nat Methods,2011,8(5):430-435.
[12] MACLEAN B,TOMAZELA D M,SHULMAN N,et al.Skyline:An open source document editor for creating and analyzing targeted proteomics experiments[J].Bioinformatics,2010,26(7):966-968.
[13] BRUSNIAK M Y,KWOK S T,CHRISTIANSEN M,et al.ATAQS:A computational software tool for high throughput transition optimization and validation for selected reaction monitoring mass spectrometry [J].BMC Bioinformatics,2011,12:78.
[14] LANGE V,MALMSTROM JA,DIDION J,et al.Targeted quantitative analysis of streptococcus pyogenes virulence factors by multiple reaction monitoring[J].Mol Cell Proteomics,2008,7(8):1 489-1 500.
[15] MARTIN D B,HOLZMAN T,MAY D,et al.MRMer,an interactive open source and crossplatform system for data extraction and visualization of multiple reaction monitoring experiments[J].Mol Cell Proteomics,2008,7(11):2 270-2 278.
[16] 卫军营,张养军,赵 炎,等.质谱智能选择反应检测用于蛋白质绝对定量中母离子对确证的方法研究[J].分析化学,2012,40(1):59-65.
[17] SONG Y,HAO Y,SUN A,et al.Sample preparation project for the subcellular proteome of mouse liver [J].Proteomics,2006,19(6):5 269-6 277.
[18] WANG G,WU W W,ZENG W,et al.Labelfree protein quantification using LC-coupled ion trap or FT mass spectrometry:Reproducibility,linearity,and application with complex proteomes[J].J Proteome Res,2006,5(5):1 214-1 223.
[19] COLAERT N,VANDEKERCKHOVE J,GEVAERT K,et al.A comparison of MS2-based label-free quantitative proteomic techniques with regards to accuracy and precision [J].Proteomics,2011,11(6):1 110-1 113.
[20] AMERICA A H,CORDEWENER J H,Comparative LC-MS:A landscape of peaks and valleys[J].Proteomics,2008,8(4):731-749.
[21] VALOT B,LANGELLA O,NANO E,et al.MassChroQ:A versatile tool for mass spectrometry quantification [J].Proteomics,2011,11(17):3 572-3 577.
猜你喜欢
中国钢铁业(2022年8期)2022-12-21
中国钢铁业(2022年7期)2022-12-21
分析仪器(2022年5期)2022-10-14
中国石油大学学报(自然科学版)(2022年4期)2022-09-05
纺织标准与质量(2022年1期)2022-07-12
成都信息工程大学学报(2021年5期)2021-12-30
化工管理(2021年7期)2021-05-13
中学生数理化·高一版(2019年12期)2019-12-31
科教导刊·电子版(2019年12期)2019-06-12
艺术探索(2019年1期)2019-04-17
