
具抗病毒活性的金刚烷胺衍生物的研究进展
2013-03-02 08:06:12沈阳化工大学制药工程教研室沈阳110142
中国现代应用药学 2013年5期
(沈阳化工大学制药工程教研室,沈阳 110142)
具抗病毒活性的金刚烷胺衍生物的研究进展
辛建创,刘 丹*,王 趱(沈阳化工大学制药工程教研室,沈阳 110142)
目的 介绍M2离子通道抑制剂金刚烷胺衍生物的研究进展,为设计新的抗流感病毒药物提供相关依据和信息。方法 对近年来关具抗流感病毒活性金刚烷胺衍生物、类似物的相关文献进行综述。结果 国内外课题组主要集中在金刚烷的1位和2位修饰,合成一系列金刚烷胺衍生物或结构类似物,并进行抗流感病毒活性测试。结论 部分金刚烷胺衍生物具有较好的抗流感病毒活性,其中一些化合物的活性优于金刚烷胺和金刚乙胺,这些结果对设计新的抗流感病毒药物具有较好的参考价值。
金刚烷胺衍生物;流感病毒;M2离子通道抑制剂
流感是一种主要通过呼吸道传播的高度传染性疾病,长期以来一直严重威胁人类健康,特别是近年来禽流感的爆发,使抗流感病毒药物的研究更具现实意义和紧迫性。目前,抗流感病毒药物主要包括盐酸阿比朵尔、金刚烷胺、金刚乙胺、扎那米韦和奥司他韦。其中金刚烷胺和金刚乙胺均属于M2离子通道抑制剂,只具有抗流感A型病毒的活性,而对B型无效[1-2]。金刚烷胺类药物对神经系统毒副作用大、易产生耐药性[3-5],为了提高其抗病毒活性,降低其不良反应,研究人员对金刚烷胺和金刚乙胺进行了大量的结构优化或改造,本文将金刚烷胺衍生物结构类型分为3类,第Ⅰ类:金刚烷2位取代的化合物;第Ⅱ类:金刚烷1位取代的化合物;第Ⅲ类:金刚烷1, 2位取代的化合物。
1 金刚烷2位取代的化合物
1.1 3-(2-金刚烷基)吡咯烷化合物
Stamatiou等[6]以金刚烷醇和2-吡咯烷酮为原料合成了目标化合物1a~1h,其结构式见图1,其中1a,1f,1h抗病毒活性(对H2N2的MIC50分别为0.60,0.38和1.7 µmol·L-1)比金刚烷胺(金刚烷胺对H2N2的MIC50为2.6 µmol·L-1)高2~7倍,化合物1a,1f的选择指数约为210。1f,1h结构中有2个氨基,推测可能存在与M2蛋白的结合位点,对抗病毒活性有利。将1a的N烷基化,得化合物1b~1e,抗病毒活性与1a相比显著降低,可能由于烷基空间位阻防碍与M2蛋白的结合。
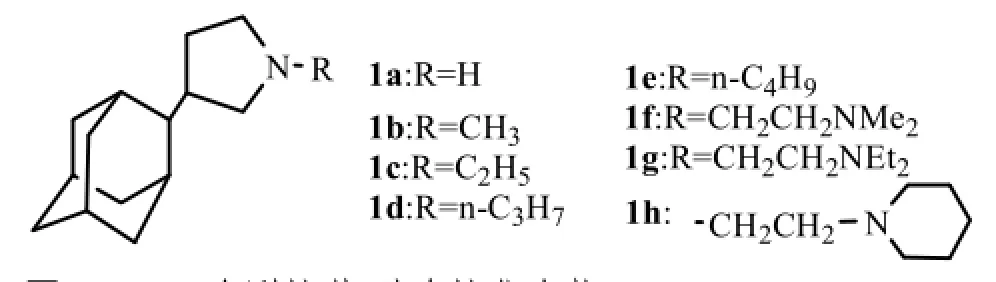
图1 3-(2-金刚烷基)吡咯烷化合物Fig1 3-(2-adamantyl) pyrrolidines
1.2 螺[N杂环-2,2’-金刚烷]化合物
Kolocouris等合成了一系列螺[N杂环-2,2’-金刚烷]化合物2.1~2.11[7-11],其结构式见图2,发现化合物2.1b抗流感A型病毒的MIC50为0.56 μg·mL-1,比金刚烷胺(MIC50为100 μg·mL-1)高178倍[7]。2.2a和2.2b抗A型流感病毒H2N2的MIC50分别为0.24 μg·mL-1和0.58 μg·mL-1,活性优于金刚烷胺(MIC50为0.8 μg·mL-1),将2.2a和2.2b哌啶环4位亚甲基用O原子取代得2.3a和2.3b也具有抗病毒活性,但抗H2N2活性比金刚烷胺弱(MIC50分别为1.7,4.3 μg·mL-1)[8]。
将2.2a和2.2b的哌啶环4位亚甲基用NH或NHCH3取代得哌嗪衍生物2.4a~2.4c,抗H3N2活性均低于金刚烷胺,(2.4a~2.4c的EC50分别为8.58,45.6 μmol·mL-1和>100 μmol·mL-1,金刚烷胺的EC50为3.35 μmol·mL-1。由此可见,在2.2a的哌啶环上再引入一个N原子,对抗A型流感病毒活性不利,N原子甲基化降低抗A型流感病毒活性[9]。
为了提高抗病毒活性,Kolocouris科研组在2.1a和2.1b的吡咯环上引入一个甲基,结果表明吡咯环上的甲基从C-3到 C-5位置上,活性逐渐增强,但是,所有的吡咯烷衍生物的抗病毒活性皆低于金刚烷胺,选择性指数也都比金刚烷胺低[10]。
以A型流感病毒H3N2评价化合物2.1a的杂环扩环和缩环类似物2.8a~2.11b的活性,。结果表明,将六元环、四元环用三元环代替会降低对A型流感病毒的活性,其中哌啶衍生物2.11a(EC50为0.16 μmol·mL-1)活性最强,是金刚烷胺的12倍,是金刚乙胺的2倍。另外N原子引入甲基会使抗A型流感病毒活性降低[11]。
遗憾的是比较以上化合物的抗流感病毒的活性具有一定的难度,因为这些化合物的合成在不同的年代,活性检测使用的A型流感病毒也不相同[12]。如1996报道了化合物2.2a和2.2b的合成,以A2/Japan,H2N2亚型评价其活性[8],2007年报道了化合物2.11a~2.11b的合成,以A/Hong Kong/7/87,H3N2亚型检测活性[11]。另外,抗病毒活性的描述也不十分准确,如化合物2.1b只报道其抗A型流感病毒,并未指明亚型[7],由于金刚烷胺对A型流感病毒不同的亚型活性不同,因此很难定量的比较所有这些化合物的抗病毒活性。

图2 螺[N杂环-2,2’-金刚烷]化合物Fig2 Spiro [azacyclo -2, 2’-adamantanes]
1.3 2-氨基(或羟基或氟)金刚烷化合物
Kolocouris等研究发现抗流感A型病毒活性较好的化合物2.1b,2.2a,2.2b(对H2N2的MIC50分别为0.56,0.24,0.58 μg·mL-1),为了评价该类化合物与M2离子通道的结合能力及抗流感病毒活性,将2.2a,2.2b结构简化,得2-氨基金刚烷4a和2-烷基-2-氨基金刚烷化合物4b~4d[13]。研究结果表明,氨基由金刚烷的1位转移到2位时,与M2蛋白结合能力减弱(金刚烷胺和4a与M2蛋白结合常数Kd分别为0.3,2.36 µmol·L-1),抗流感病毒活性逐渐降低(金刚烷胺和4a对H2N2的EC50分别为1.1,2.8 µmol·L-1)。当2-金刚烷胺4a的2位连有不同的烷基时(4b~4d),随着可旋转的直链烷基增大,与M2离子通道结合能力逐渐减弱(与M2蛋白结合常数Kd分别为3.60,6.70, 8.71 µmol·L-1),但抗病毒活性却逐渐提高(4b,4c,4d 的EC50分别为3.5,<1.9,<1.7 µmol·L-1)。与4b,4c,4d相比,2位连有刚性哌啶环的化合物2c与M2离子通道结合能力增强(Kd为0.39 µmol·L-1),刚性哌啶环位于M2受体的亲脂口袋,比能自由旋转的烷基具有更合理的取向,其抗病毒活性(EC50为1.0 µmol·L-1)高于化合物4b,4c和4d,与金刚烷胺活性相当。
氨基金刚烷类化合物的氨基通过氢键作用和与M2蛋白结合,以羟基或氟原子替换4b的氨基得化合物3a和5a,3a失去与M2的结合能力,5a与M2的结合能力显著降低(Kd>26 µmol·L-1),但3a的抗病毒活性(EC50为3.0 µmol·L-1)与4b(EC50为3.5 µmol·L-1)相当,且没有拮抗N-甲基-D-天门冬氨酸(NMDA)的作用,作为抗流感病毒化合物值得进一步研究。研究结果也说明化合物与M2离子通道相结合能力并不与其抗病毒活性成正相关。
2-氨基(或羟基或氟)金刚烷化合物结构式见图3。
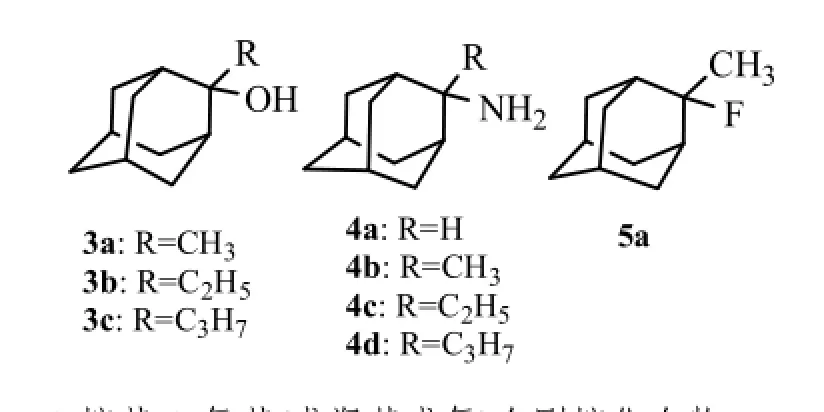
图3 2-烷基-2-氨基(或羟基或氟)金刚烷化合物Fig3 2-ankyl-2-amino(or hydroxy, fluoride) adamantanes
2 1-金刚烷基衍生物
2.1 2-(1-金刚烷基)-N-杂环烷烃化合物
Stamatiou等[14]合成2-(1-金刚烷基)吡咯烷化合物6a,6b。2-(1-金刚烷基)哌啶化合物7a~7f以及2-(1-金刚烷基)吖庚因化合物8a~8c。6a和7a对抗H2N2病毒活性(EC50分别为2.1,3.3 µmol·L-1)分别是金刚烷胺(EC50为45 µmol·L-1)的18倍和14倍,是金刚乙胺(EC50为13.9 µmol·L-1)的6倍和4倍,而化合物8a(EC50为189 µmol·L-1)活性较差,由此可见,金刚烷1位所连环的大小影响抗病毒活性,由五元环到七元环,活性依次降低。另外化合物6a和7a的N烷基化后活性显著降低(6b和7b 的EC50分别为196,978 µmol·L-1),只有7a的N,N-二甲基氨乙基取代物7e (EC50为51.3 µmol·L-1)与金刚烷胺活性相当,可能存在3个药效团:金刚烷基及2个氨基。结构式见图4。
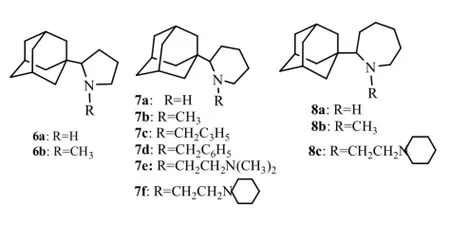
图4 2-(1-金刚烷基)-N-杂环烷烃化合物Fig4 2-(1-adamantyl)-azacyclanes
Zoidis等[15]合成了2-(1-金刚烷基)-2-甲基-吡咯9a和9b,2-(1-金刚烷基)-2-甲基-氮杂环丁烷10a和10b,以及2-(1-金刚烷基)-2甲基-氮杂环丙烷11a和11b,其结构式见图5。H2N2病毒的活性最强(EC50为1.56 µmol·L-1),分别是金刚乙胺(EC50为14 µmol·L-1)的9倍和金刚烷胺(EC50为42 µmol·L-1)的27倍,10a的EC50为1.96 µmol·L-1,抗病毒活性也优于金刚烷胺和金刚乙胺。将五元环和四元环用三元环代替,降低了抗A型流感病毒的活性,如11a的EC50为>55 µmol·L-1。9a,10a,11a的N甲基化产物9b,10b,11b的EC50分别为>194,4.1 µmol·L-1和>259 µmol·L-1,可见杂环N原子上引入甲基对抗病毒活性不利。
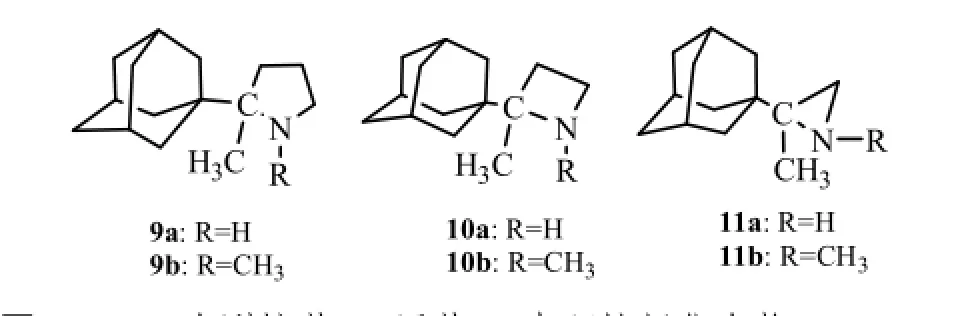
图5 2-(1-金刚烷基)-2-甲基-N-杂环烷烃化合物Fig5 2-(1-adamantyl)-2-methyl-azacyclanes
Tataridi等[16]报道了金刚乙胺12的类似物12a~15c,该类化合物特点是向金刚乙胺的药效团1-氨乙基上引入第二个氨基得1,2-二氨基乙基取代物。当金刚乙胺12(EC50为19.1 µmol·L-1)的α-甲基被α-氨甲基取代得12a(EC50为18.3 µmol·L-1),抗A型H3N2病毒活性与金刚乙胺比略有提高;化合物7a的哌啶环4-CH2被NH取代得15a,15a活性(EC50为24.1 µmol·L-1)高于7a(EC50为70.5 µmol·L-1)。12a和15a结构中均有2个氨基,可能与M2蛋白形成2个氢键。13a和14a分别为具有羰基的五元和六元N-杂环衍生物,EC50分别为72.6和91.8 µmol·L-1,结构中也有2个氨基,可以与M2蛋白形成2个氢键,但其活性均低于12a和15a,可能由于羰基的存在,使得药物分子与M2蛋白无法采取合适的取向,进而无法相互契合,发生相互作用。将12a~15a的N甲基化,抗H3N2病毒活性不同程度降低,甚至失去活性,如13b,14b,15b,15c的EC50分别为269,604,197,351 µmol·L-1。这与文献[14-15]报道的结果是一致的。结构式见图6。
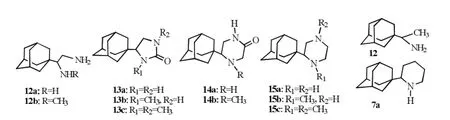
图6 金刚乙胺类似物Fig6 Rimantadine analogs
2.2 1-金刚烷基唑类化合物
Zarubaev等[17]合成了一系列金刚烷的唑类衍生物,对这些化合物进行抗流感病毒活性(A型/Puerto Rico/8/34)检测,结果显示(1-金刚烷基)二唑和(1-金刚烷基)-1,2,4-三唑(16a~17b)没有表现出很高的抗病毒活性。在金刚烷基3位上引入一个氨乙基可略微提高1,2,4三唑的抗病毒活性,如21c的EC50为15 µmol·L-1,17b的EC50为 59 µmol·L-1。金刚烷胺四唑类衍生物中,当金刚烷基3位有氨基或氨乙基取代时,活性相应提高,22a和22b(EC50分别为22,13 µmol·L-1),23a和23b (EC50分别为23,8 µmol·L-1),而18a和18b(EC50分别为56,77 µmol·L-1)。值得注意的是金刚烷基在四唑环的位置对活性影响较大,如18a 的活性(EC5056 µmol·L-1)相对 19a(EC5011 µmol·L-1)降低5倍。结构式见图7。
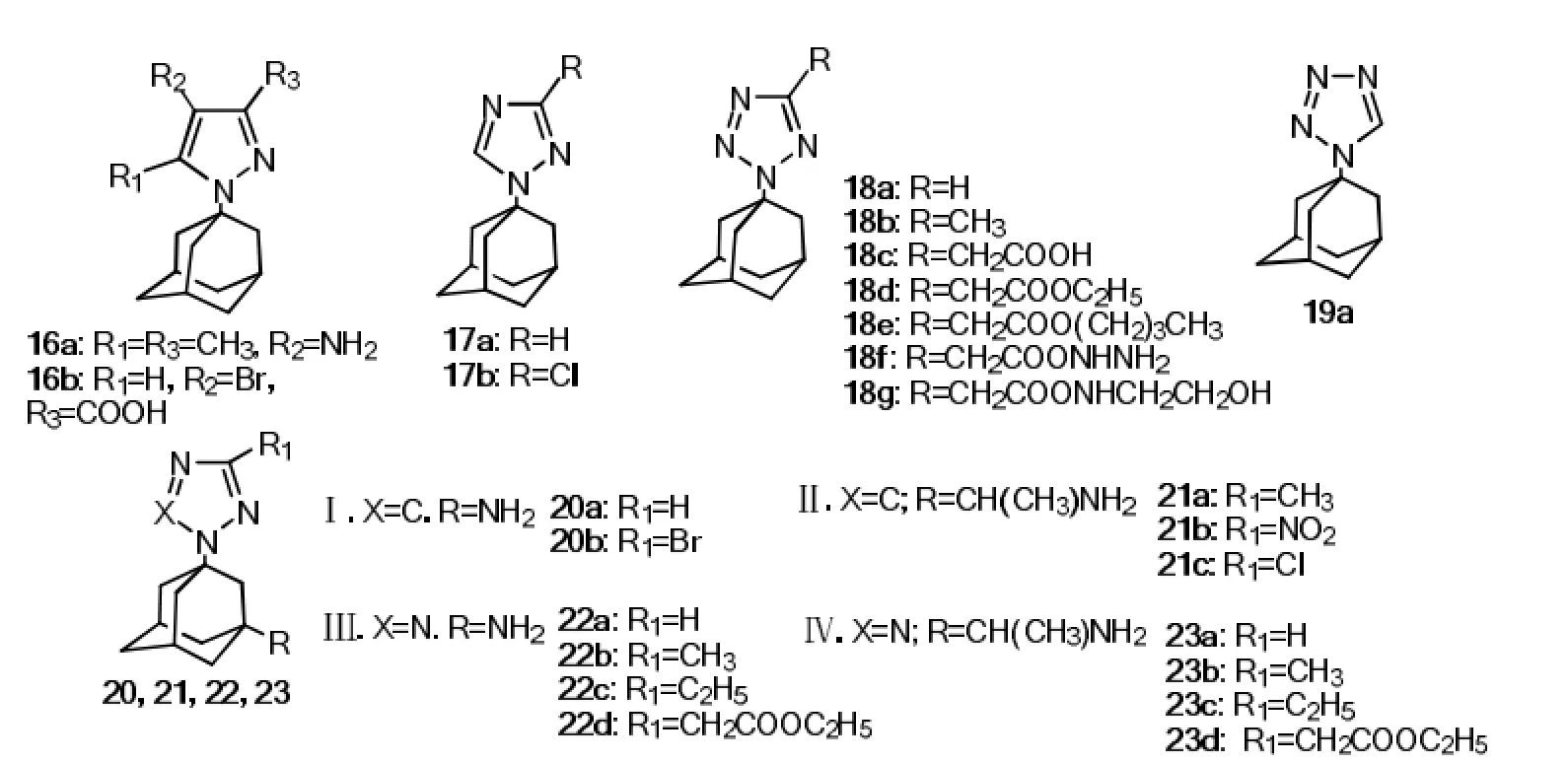
图7 (1-金刚烷基)唑类化合物Fig7 (1-adamantyl)-ozazoles
化合物结构中包含2个四唑环或者2个金刚烷基,见图8,将失去抗病毒活性(24a EC50>500 µmol·L-1,24b EC50为180 µmol·L-1,27a EC50为57 µmol·L-1)。
以硫代四唑环取代异硫代氰基,抗病毒活性提高,(如25c vs 25b,EC50分别为8和150 µmol·L-1);25a的金刚烷基3位引入羟基,抗病毒活性提高(25c vs 25a,EC50分别为8和15 µmol·L-1)。当四唑环上连有苯基,活性降低(26a vs 26b,EC50分别为130和3 µmol·L-1)。结构式见图9。
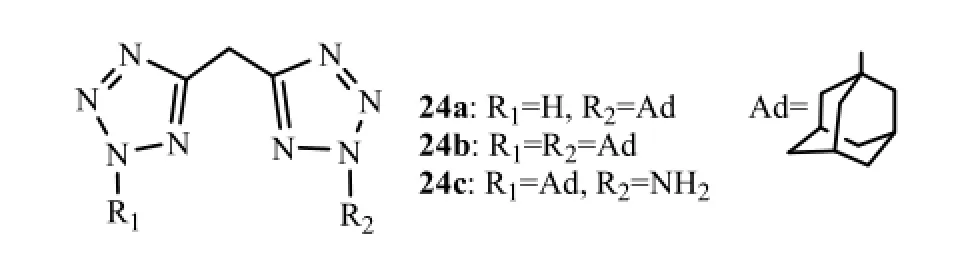
图8 含2个四唑环或两个金刚烷基的化合物Fig8 Diadamanthyl and ditetrazole derivatives
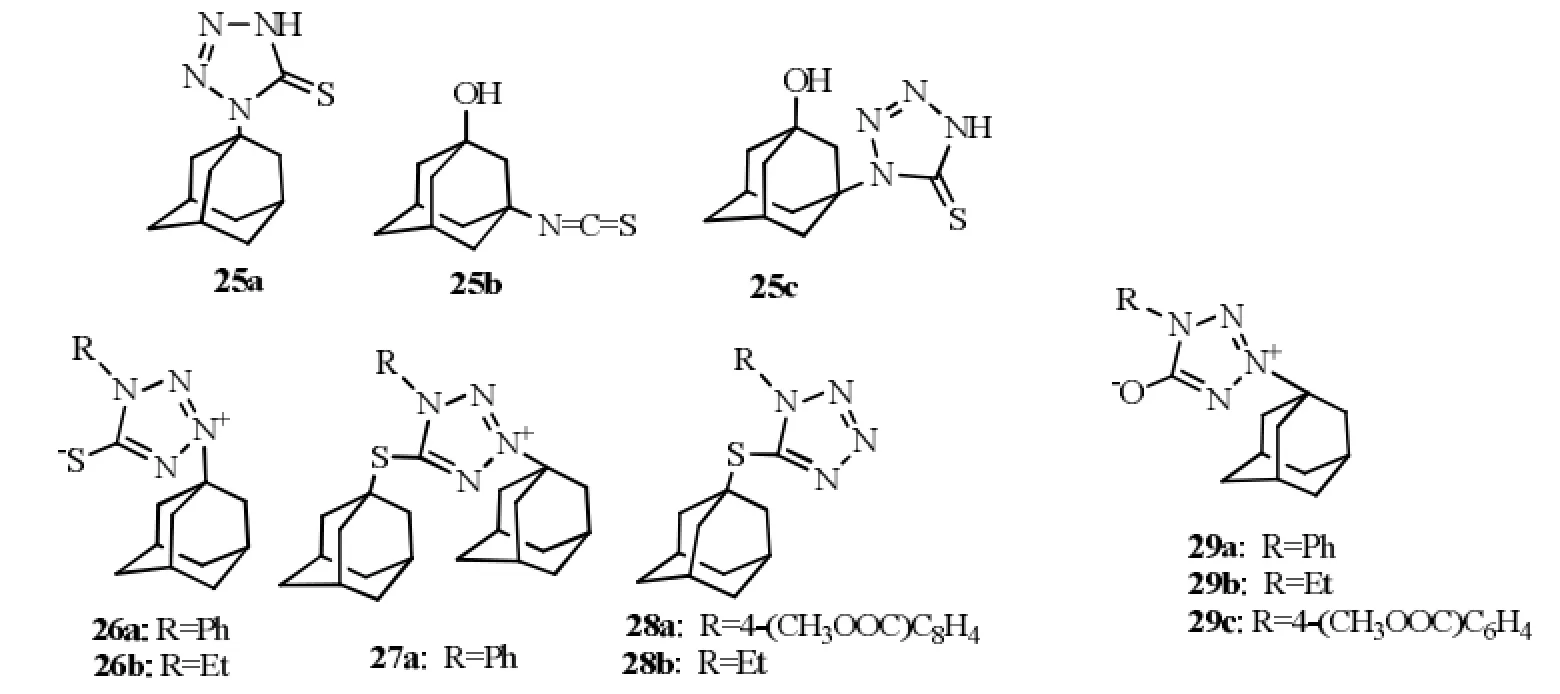
图9 (1- 金刚烷基)-硫代(或氧代)四唑类化合物Fig9 (1-adamantyl)-thio(or oxo) tetrazoles
2.3 金刚烷胺氨基酸衍生物
黎万等[18]以金刚烷甲酰氯和氨基酸甲酯盐酸盐为起始原料,经酰化、水解、成盐等反应得到金刚烷胺衍生物(30a~30h,31a~31d),其结构式见图10。以金刚烷胺为阳性对照,采用幼犬肾(MDCK)细胞系噬斑形成实验测定目标化合物的抗禽流感病毒活性。结果显示,仅有化合物30d显示出较好的抗禽流感病毒活性,EC50为6.67 mg·L-1。
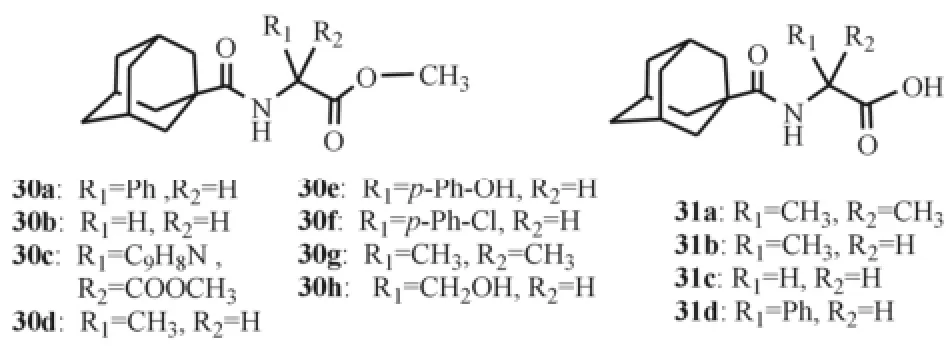
图10 金刚烷甲酰氯和氨基酸反应得到的衍生物Fig10 Amantadine derivatives obtained from 1-admantanecarbonnyl chloride and amino acids
3 1,2-金刚烷基杂环化合物
2008年,Zoidis等[19]合成了1,2金刚烷基吡咯烷类似物32a~34a,结构式见图11。结果发现化合物32a,33a,34a抗A型病毒活性显著(EC50分别为2.2,0.46,1.1 µmol·L-1)。33a活性最强,是金刚烷胺(EC50为2.0 µmol·L-1)的4倍,与金刚乙胺(EC50分别为0.36 µmol·L-1)活性相当。N烷基化后,活性皆低于无取代基的化合物。N原子与金刚烷基1位之间的距离和抗病毒活性密切相关(32a vs 33a)。
2009年,Zoidis等[20]在文献[19]的基础上,合成了1,2金刚烷基哌啶化合物35a~37b,结构式见图12。同样哌啶环上N原子与金刚烷基1位间的距离不同会导致抗病毒活性不同,如35a EC50为4.1 µmol·L-1,37a活性最强,EC50为0.6 µmol·L-1,而36a在最大检测浓度(500 µmol·L-1)时无活性。哌啶环上N烷基化后,活性降低,如37a EC50为0.6 µmol·L-1,37b在最大检测浓度(500 µmol·L-1)时无活性(37a vs 37b)。
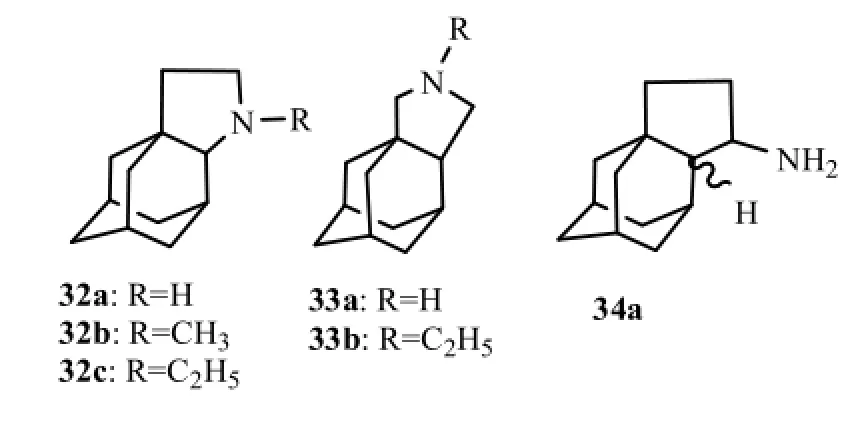
图11 1,2金刚烷基吡咯烷类似物Fig11 1, 2-annulated adamantanopyrrolidines
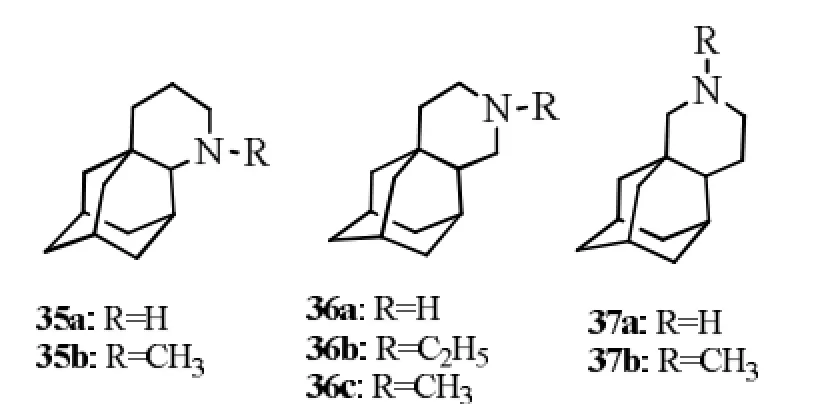
图12 1,2金刚烷基哌啶化合物Fig12 1, 2-annulated adamantane piperidines
4 讨论
M2离子通道蛋白抑制剂是最早上市的抗流感药物,但其神经毒性、长期服用易产生耐药毒株和对B型流感无效等缺点阻碍了其在临床中的广泛应用。近10年里,新的金刚烷胺类似物不断被合成出来,这些衍生物保持了金刚烷胺、金刚乙胺的药效团,其中一些化合物抗病毒活性甚至优于金刚烷胺和金刚乙胺。这些衍生物对A型流感病毒显示出很强的抑制活性,其结构的共同特点是碳环或杂环都与金刚烷的基本骨架相连。多数情况下,杂环上N原子甲基化后会降低衍生物抗病毒活性。也有研究者用固态NMR、液相NMR、X-射线晶体衍射以及分子动力模拟计算等方法来研究M2离子通道蛋白的结构,以阐明M2离子通道的组成、离子通道传递机制及抗药机制[21-24]。目前虽然未有新的M2离子通道抑制剂上市,但这些研究结果对设计新型抗流感病毒药物的推动作用毋容置疑。
REFERENCES
[1] YIN L, JIANG J D. Research progress of anti-influenza virus agents [J]. Chin New Drugs J(中国新药杂志), 2004, 13(8): 685-687.
[2] ZHANG Q, ZHAO R S, XIONG R S, et al. Research progress of anti-influenza virus agents [J]. Acta Pharm Sin(药学学报), 2010, 45(3): 289-299.
[3] O YLX, WAN X Y, BI S Y. Amantadine [J]. Prog Japan Med (日本医学介绍), 2004, 25(7): 307-309.
[4] WEINSTOCK D M, ZUCCOTTI G. Adamantane resistance in influenza A [J]. JAMA, 2006, 295(8): 934-936.
[5] SAITO R, LI D J, SUZUKI H. Amantadine-resistant influenza A(H3N2)virus in Japan, 2005-2006 [J]. N Engl J Med, 2007, 356(3): 312-313.
[6] STAMATIOU G, KOLOCOURIS A, KOLOCOURIS N, et al. Novel 3-(2-adamantyl) pyrrolidines with potent activity against inf l uenza A virus - identif i cation of aminoadamantane derivatives bearing two pharmacophoric amine groups [J]. Bioorg Med Chem Lett, 2001, 11(16): 2137-2142.
[7] KOLOCOURIS N, FOSCOLOS G B, KOLOCOURIS A, et al. Synthesis and antiviral activity evaluation of some aminoadamantane derivatives [J]. J Med Chem, 1994, 37(18): 2896-2902.
[8] KOLOCOURIS N, KOLOCOURIS A, FOSCOLOS G B, et al. Synthesis and antiviral activity evaluation of some aminoadamantane derivatives. 2 [J]. J Med Chem, 1996, 39(17): 3307-3318.
[9] FYTAS C, KOLOCOURIS A, FYTAS G, et al. Influence of an additional amino group on the potency of aminoadamantanes against influenza virus A. II-Synthesis of spiropiperazines and in vitro activity against influenza A H3N2 virus [J]. Bioorg Chem, 2010, 38(6): 247-251.
[10] STYLIANAKIS I, KOLOCOURIS A, KOLOCOURIS N, et al. Spiro [pyrrolidine-2, 20-adamantanes]: synthesis, anti-influenza virus activity and conformational properties [J]. Bioorg Med Chem Lett, 2003, 13(10): 1699-1703.
[11] KOLOCOURIS N, ZOIDIS G, FOSCOLOS G B, et al. Design and synthesis of bioactive adamantane spiro heterocycles [J]. Bioorg Med Chem Lett, 2007, 17(15): 4358-4362.
[12] DUQUE M D, TORRES E, VALVERDE E, et al. Inhibitors of the M2 channel of influenza A virus [M]. Kerala: Transworld Research Network, 2011: 35-64.
[13] KOLOCOURIS A, SPEARPOINT P, MARTIN S, et al. Comparisons of the inf l uenza virus A M2 channel binding aff i nities, anti-inf l uenza virus potencies and NMDA antagonistic activities of 2-alkyl-2-aminoadamantanes andanalogues [J]. Bioorg Med Chem Lett, 2008, 18(23): 6156-6160.
[14] STAMATIOU G, FOSCOLOS G B, FYTAS G, et al. Heterocyclic rimantadine analogues with antiviral activity [J]. Bioorg Med Chem, 2003, 11(24): 5485-5492.
[15] ZOIDIS G, FYTAS C, PAPANASTASIOU I, et al. Heterocyclic rimantadine analogues with antiviral activity [J]. Bioorg Med Chem, 2006, 14(10): 3341-3348.
[16] TATARRIDIS D, FYTAS G, KOLOCOURIS A, et al. Inf l uence of an additional 2-amino substituent of the 1-aminoethyl pharmacophore group on the potency of rimantadine against inf l uenza virus A [J]. Bioorg Med Chem Lett, 2007, 17(3): 692-696.
[17] ZARUBAEV V V, GOLOD E L, ANFIMOV P M, et al. Synthesis and anti-viral activity of azolo-adamantanes against inf l uenza A virus [J]. Bioorg Med Chem, 2010, 18(2): 839-848.
[18] LI W, LV L, QU J, et al. Designed and synthesized of amantadine derivstives and their anti-avian influenza virus activity [J]. Chin J Med Chem (中国药物化学杂志), 2011, 21(5): 345-351.
[19] ZOIDIS G, TSOTINIS A, KOLOCOURIS N, et al. Design and synthesis of bioactive 1, 2-annulated adamantane derivatives [J]. Org Biomol Chem, 2008, 6(17): 3177-3185.
[20] ZOIDIS G, KOLOCOURIS N, NAESENS L, et al. Design and synthesis of 1, 2-annulated adamantane piperidines with anti-inf l uenza virus activity [J]. Bioorg Med Chem, 2009, 17(4): 1534-1541.
[21] CADY S D, WANG J, WU Y B, et al. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy [J]. J Am Chem Soc, 2011, 133(12): 4274-4284.
[22] KOLOCOURIS A, HANSEN R K, BROADHURST R W. Interaction between an amantadine analogue and the transmembrane portion of the influenza A M2 protein in liposomes probed by1H NMR spectroscopy of the ligand [J]. J Med Chem, 2004, 47(20): 4975-4978.
[23] SCHELL J R, CHOU J J. Structure and mechanism of the M2 Proton channel of influenza A virus. [J]. Nature, 2008, 451(7178): 591-595.
[24] STOUFFER A L, ACHARYA R, SALOM D, et al. Structural basis for the function and inhibition of an influenza virus proton channel [J]. Nature, 2008, 451(7178): 596-599.
Progress of Amantadine Derivatives with Antiviral Activity
XIN Jianchuang, LIU Dan*, WANG Zan(Faculty of Pharmaceutical Engineering, Shenyang University of Chemical Technology, Shenyang 110142, China)
OBJECTIVE To review the research advances in amantadine derivatives with antiviral activity and to provide ideas for research and development of new anti-influenza and anti-virus drug. METHODS Review related literatures about the recent research progress on amantadine derivatives and analogs. RESULTS Many derivatives and analogs of amantadine were synthesized and their anti-influenza and anti virus activities were evaluated. The structural modification focused on the position 1 and position 2 of admantane. CONCLUSION Several active compounds are found, some of which have more potency than amantadine and rimantadine.
adamantine derivatives; influence avirus; M2 proton channel inhibitor
R961
A
1007-7693(2013)05-0552-07
2012-08-11
辛建创,男,硕士生 Tel: (024)89383903 E-mail: xjc1333399@163.com*
刘丹,女,博士,副教授 Tel: (024)89383903 E-mail: liudan20040318@163.com
猜你喜欢
中国康复(2022年8期)2022-08-31 07:14:56
中国渔业质量与标准(2022年3期)2022-07-23 01:52:46
中国康复(2022年11期)2022-03-02 11:20:38
——以塔里木盆地塔中地区凝析油为例
石油实验地质(2021年5期)2021-11-01 06:55:44
云南化工(2021年2期)2021-05-06 01:06:20
地球科学与环境学报(2020年2期)2020-03-26 10:01:28
石油与天然气地质(2018年4期)2018-08-01 06:36:56
结核与肺部疾病杂志(2015年2期)2015-07-18 11:00:27
食品工业科技(2013年12期)2013-08-07 09:14:36
心脑血管病防治(2011年3期)2011-09-15 08:18:48
