
N-杂环卡宾催化的成环反应研究进展
2013-03-02 08:06:13浙江医学高等专科学校药学系杭州310053
中国现代应用药学 2013年5期
(浙江医学高等专科学校药学系,杭州 310053)
N-杂环卡宾催化的成环反应研究进展
倪佳婷,董 晶,胡文君,杜文婷*(浙江医学高等专科学校药学系,杭州 310053)
目的 介绍N-杂环卡宾催化的成环反应研究进展。方法 综合国内外报道的文献,阐述N-杂环卡宾催化的成环反应。结果 列出了N-杂环卡宾催化的分子内Stetter反应、亲核取代反应、环加成反应和串联反应等成环方法,重点分析了N-杂环卡宾催化的[4+2],[3+2],[2+2]和[3+3]环加成合成法。结论 N-杂环卡宾催化成环反应的发展在有机合成方法学中为高效成环提供了新的途径和策略。
N-杂环卡宾(NHC);成环反应;催化
自1991年Arduengo等[1]成功得到首个稳定的N-杂环卡宾-咪唑-2-碳烯,N-杂环卡宾化学成为有机化学家争相研究的热点领域,尤其是作为一种亲核性的有机小分子催化剂。N-杂环卡宾的独特性之一在于它可以催化许多羰基化合物发生极性反转,产生的具有亲核性的酰基阴离子中间体,可以与多种亲电底物反应,广泛的用于有机合成反应,成为一种有效的合成策略。近年来,通过N-杂环卡宾催化合成环状结构化合物方法有突破性进展,所涉及的反应类型多种多样,且涉及的成环产物如β-内酯、环酮、稠环化合物等往往具有重要合成用途或重要生理活性,或是医药合成过程中的重要中间体,或是药物、活性化合物的关键药效团或结构模块。如吡啶环酮可控制人体内铁的含量,对防治老年痴呆症和地中海贫血症等有很好疗效[2],又如具有多种生物活性的天然产物Steganone结构中包含的内酯结构就可以尝试用N-杂环卡宾催化成环方法加以合成[3]。
本文就近五年来NHC催化的成环反应的各种方法及其特点进行分析讨论。
1 分子内Stetter反应
1996年,Enders等[4]首次报道了不对称分子内Stetter反应,采用手性N-杂环卡宾催化剂A,简单有效的合成手性的苯并吡喃酮衍生物,有中等的收率和ee值。之后随着越来越多手性催化剂被合成出来,通过不对称分子内Stetter反应合成环状结构化合物的方法也受到更多化学家的关注。
Rovis推动了N-杂环卡宾催化的分子内Stetter反应的发展。2002年Rovis等[5]采用新的手性三氮唑类N-杂环卡宾催化剂对不对称分子内Stetter反应进行了改进,最佳的产率和ee值分别为95%和97%。其中底物结构中的X原子可以为O,S,N,C,而且脂肪醛底物也适用该反应体系,以很好的产率和ee值得到环戊酮衍生物。2006年,Tomioka等[6]在此基础上,以手性咪唑啉类N-杂环卡宾催化剂催化合成环戊酮衍生物。见图1。
Rovis等[7]合成了一系列含有一个手性中心的外消旋脂肪族底物,用于合成2,3-和2,4-双取代的环戊酮,总收率达90~97%。通过平行动力学拆分,trans和cis的异构体均可获得很高的ee值。而2,5-双取代的环戊酮合成受底物控制,能高选择性得到cis的异构体,但只能得到外消旋体;而选择非手性的三氮唑类N-杂环卡宾催化剂时,非对映体选择性很差。笔者将其归因于手性和非手性催化剂的空间排列上有很大差别。见图2。
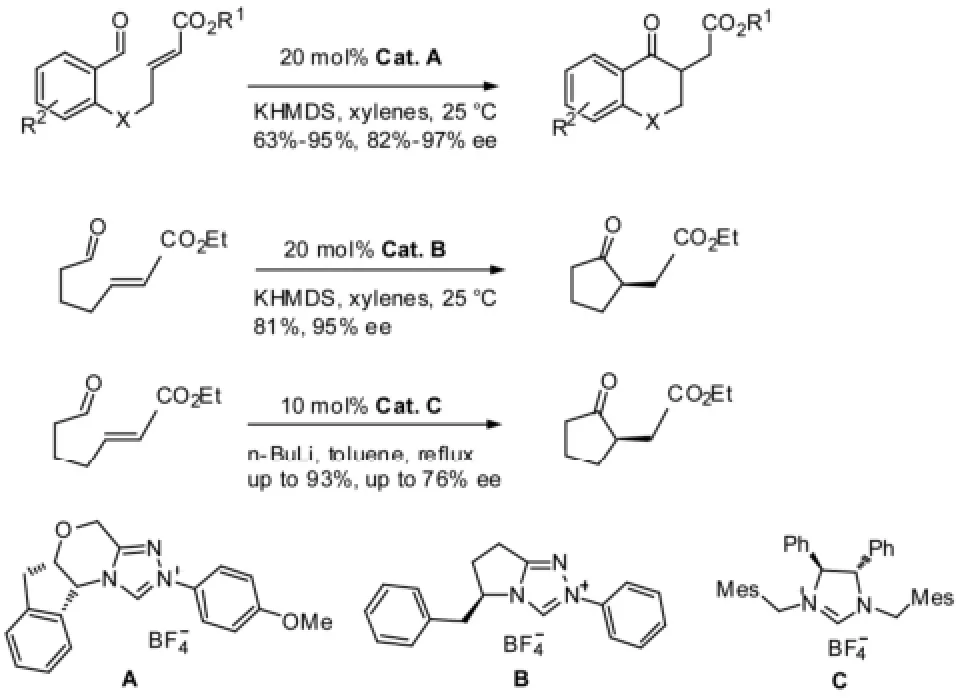
图1 分子内Stetter反应例1Fig1 Example 1 of intramolecular stetter reaction
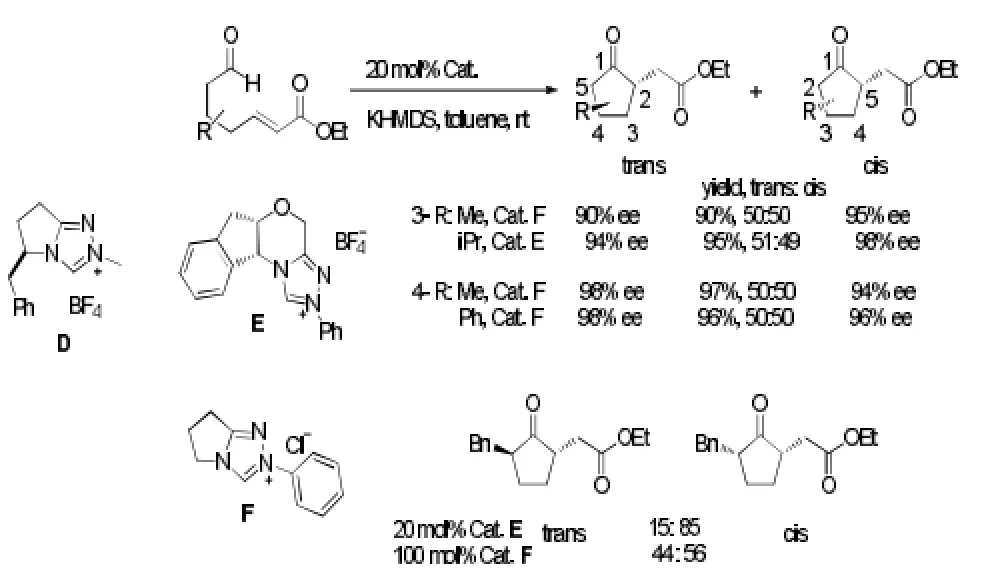
图2 分子内Stetter反应例2Fig2 Example 2 of intramolecular Stetter reaction
Hamada[8]等受到Rovis等的启发,通过非手性的噻唑类N-杂环卡宾催化分子内Stetter反应,简便高效的合成含有季碳的色满-4-酮和2,3-二氢喹啉-4-酮。他们发现,三乙胺和叔丁醇用于反应时,结果最好。而且E式和Z式的烯烃都同样适用与该反应体系。见图3。
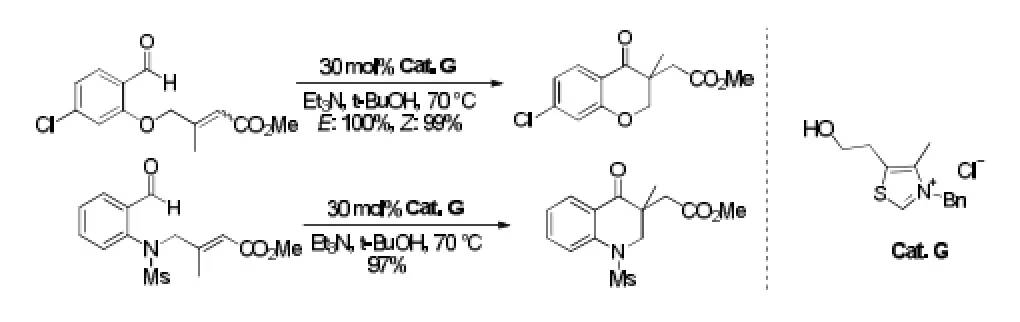
图3 分子内Stetter反应例3Fig3 Example 3 of intramolecular stetter reaction
Rovis等[9]将分子内Stetter进一步优化,成功合成出氢化苯并呋喃酮,最多可以获得3个毗连的手性中心,且产率、对映体选择性和非对映体选择性均很好。见图4。
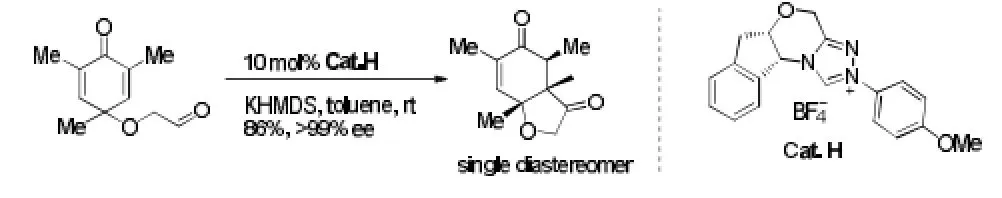
图4 分子内Stetter反应例4Fig4 Example 4 of intramolecular stetter reaction
最近,You课题组[10]以取代苯酚的去芳构化产物环己二烯酮作为Stetter反应的受体,在D-樟脑衍生的三唑类卡宾前体盐H催化下,与取代基上的醛发生分子内Stetter反应,得到手性的三元环产物。以DIEA为碱,邻二甲苯为溶剂时,有中等至较好的产率和很好的ee值。笔者指出,该催化剂的高活性是由于邻位N上的吸电子芳基取代使得催化剂上的质子具有酸性。值得一提的是,当底物为二甲基取代的环己二烯酮时,只得到单一的非对映体,ee值达到99%,但产率仅为9%。见图5。
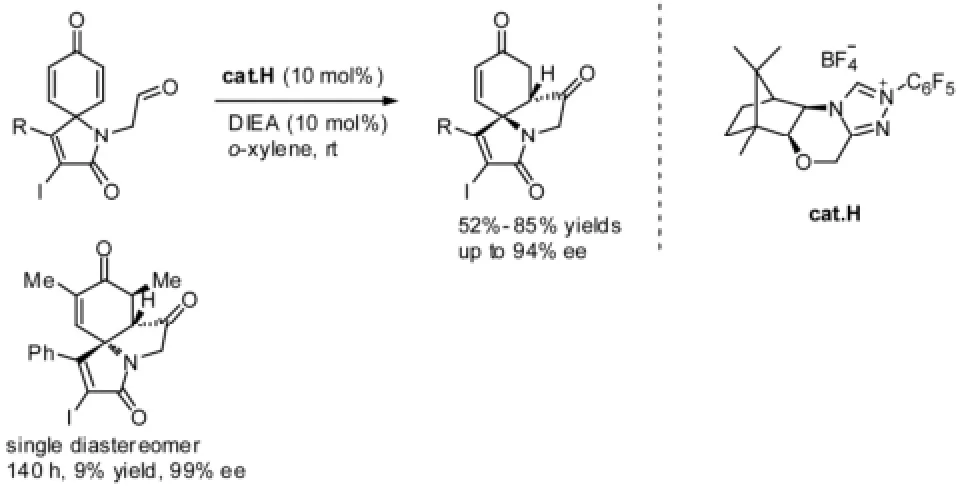
图5 分子内Stetter反应例5Fig5 Example 5 of intramolecular stetter reaction
2 亲核取代反应
N-杂环卡宾催化的亲核取代反应是近几年发展起来的,通过亲核取代反应合成环状结构化合物的报道较少。2006年Fu等[11]在研究N-杂环卡宾和钯催化剂共同催化的烷基与烯烃的Heck反应时发现,不加钯催化剂时,偶联的效果更好。因此开发出N-杂环卡宾催化的α,β-不饱和酯的β-烷化反应,可以得到四至六元环,产率从中等到很好。见图6。
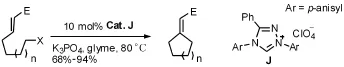
图6 亲核取代反应例1Fig6 Example 1 of nucleophilic substitution reaction
环丙烷通常都是通过α,β-不饱和醛、酮与硫叶立德或二级胺共轭加成后进行分子内闭环反应(Michael-Initiated Ring Closure,MIRC)得到。最近Nair等[12]将MIRC反应的底物拓展到氰基丙烯酸酯与α-溴苯乙酮,并首次实现以N-杂环卡宾催化MIRC反应。经过筛选,当使用DBU作碱,二氯甲烷为溶剂时,产率达94%。见图7。
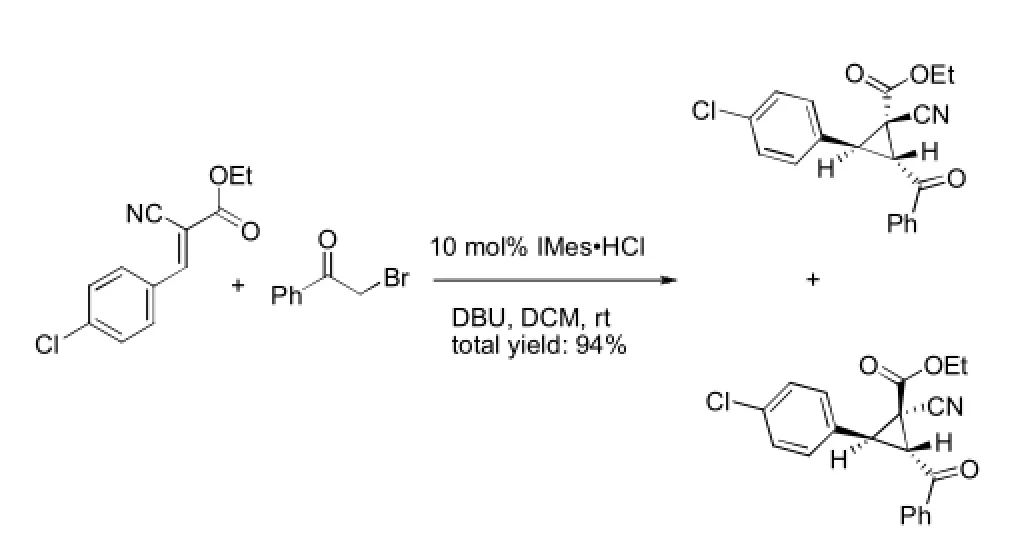
图7 亲核取代反应例2Fig7 Example 2 of nucleophilic substitution reaction
3 环加成反应
环加成反应早已被公认为是合成环状结构化合物最有效的方法,各种类型的环加成反应被广泛的用于合成五元至八元环状结构化合物,如[4+2],[3+2]等环加成反应。
3.1 [4+2]环加成
[4+2]环加成反应已经有80多年历史,它是构筑六元环最重要的方法之一。而且当引入恰当的手性底物或催化剂时,可得到高对映选择性产物。
Ye[13]报道了一系列以取代乙烯酮为亲双烯体的[4+2]环加成反应。如当K-1为催化剂,碳酸铯为碱,THF为溶剂,与α,β-不饱和酮反应,主要得到trans的δ-内酯;相同条件下,他还首次实现与N-苯甲酰二氮烯的[4+2]环加成[14],其产物的对映体选择性可以通过调整催化剂的取代基来转变。K-2用于取代乙烯酮与3-烯吲哚酮[15]的不对称成环反应,能以99%的产率得到含有吲哚基团和二氢吡喃酮的产物,有较好的ee值和dr值,开辟了一种简便、高选择性合成含吲哚基团化合物的新方法。最近Ye[16]又报道了首例乙烯酮与1-氮杂二烯的[4+2]环加成反应,使用的手性催化剂(-)F,碳酸铯为碱,苯和DME为溶剂,得到高度官能团化的3,4-二氢吡啶-2-酮,可以方便的进行结构衍生。见图8。
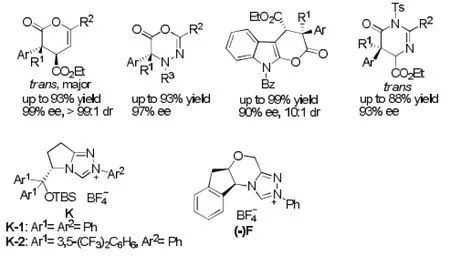
图8 [4+2]环加成反应例1Fig8 Example 1 of [4+2] cycloaddition reaction
Lupton[17]以α,β-不饱和羰基化合物为底物,经历α,β-不饱和酰基咪唑盐离子关键中间体过程,通过共轭加成、质子迁移、酰化等,得到二氢吡喃酮产物。在该反应中,只有IMes·HCl和IDip·HCl这类大位阻的咪唑类催化剂有较好的催化活性,而且只有手性催化剂G才能得到77%的收率和50%的ee值。见图9。吡喃酮产物及其中间体在医药领域中的应用正在研究中。
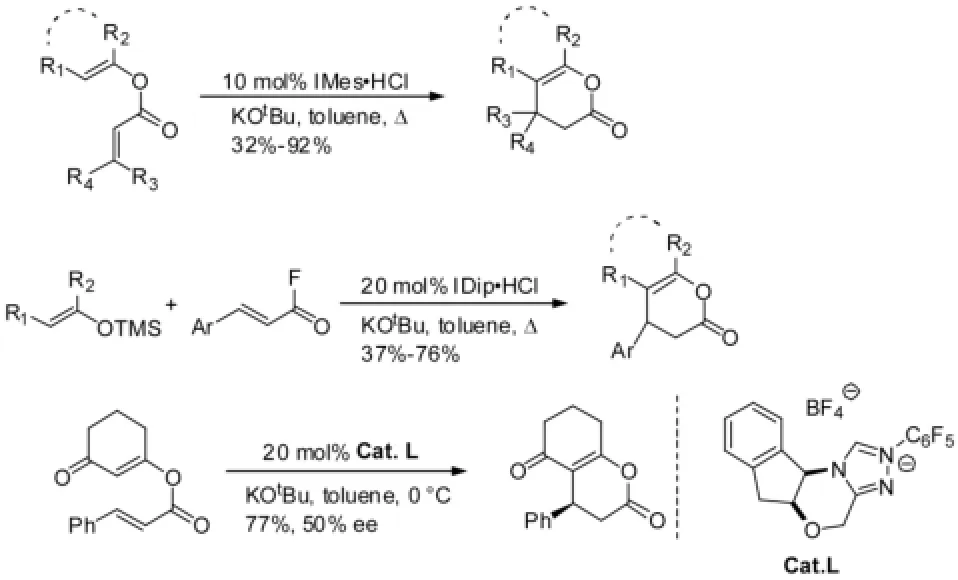
图9 [4+2]环加成反应例2Fig9 Example 2 of [4+2] cycloaddition reaction
Zhong课题组[18]对Ye课题组发展的不对称合成吲哚并二氢吡喃酮衍生物的方法进行了优化,他们以α-氯代醛为亲双烯体前体,生成的Breslow中间体消除HCl,得到烯醇负离子中间体,与2-氧代吲哚啉-3-亚基发生氧杂-Diels-Alder反应。对映体选择性和非对映体选择性都有明显的提高,不同类型的醛均只生成cis的产物,最高达到99%的产率,大于99%的ee值。笔者认为,该反应出色的非对映体选择性和绝对的立体化学来源于高度优先的endo-cis-过渡态。一方面,cis-烯醇负离子比trans-形式的热力学更稳定;另一方面,该过渡态模型中,re-面完全被催化剂中的二氢茚结构封住,只能在si-面发生[4+2]环加成。见图10。
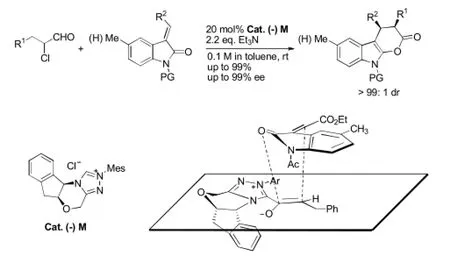
图10 [4+2]环加成反应例3Fig10 Example 3 of [4+2] cycloaddition reaction
3.2 [3+2]环加成
1,3−偶极环加成反应是合成五元环状化合物的一种重要的方法,[3+2]环加成与Diels-Alder反应类似,但是其选择性较差,因此,近年来的研究重点是对区域选择性、非对映选择性、对映选择性的控制。
Scheidt等[19]报道了在N-杂环卡宾催化下,α,β-不饱和醛与二氮烯的[3+2]环加成反应,生成单一的区位异构体吡唑啉酮。该反应的关键是要找到合适的反应底物和条件,确保卡宾优先与醛反应,而不是与具有亲电性的二氮烯发生副反应。当使用催化剂N时,有60%~84%的收率;手性催化剂可以选择性的控制产物的手性。见图11。
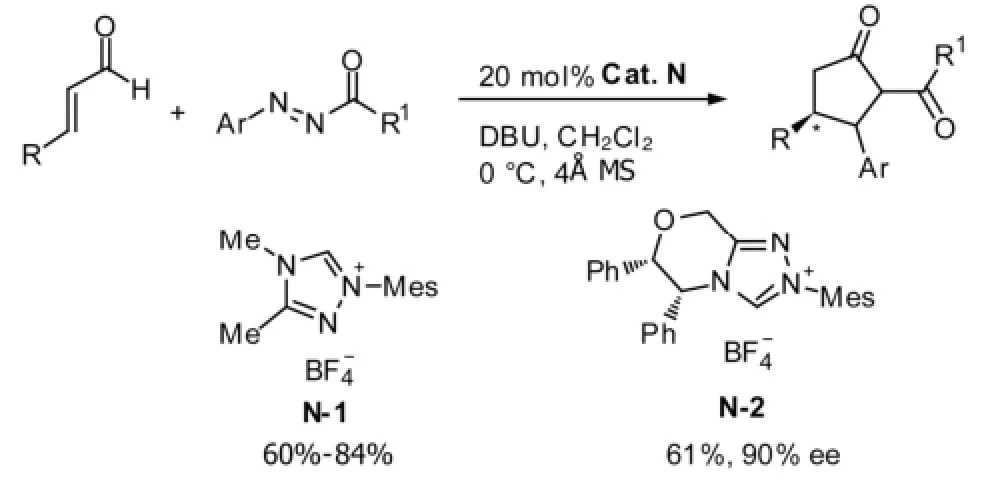
图11 [3+2]环加成反应例1Fig11 Example 1 of [3+2] cycloaddition reaction
由于α,β-不饱和醛制备的困难,大大限制了其作为1,3-偶极子的应用。Bode[20]开发出一种新型的α,β-不饱和醛的代替品——α׳-羟基-α,β-不饱和酮。此类底物不仅原料易得,合成简便、易于保存,而且可以用于N-杂环卡宾催化的成环反应。N-杂环卡宾进攻α׳-羟基-α,β-不饱和酮的羰基碳,再发生逆安息香缩合反应,离去丙酮后,与各种亲偶极子进行[3+2]环加成,脱去CO2得到相应的五元环状化合物。见图12。
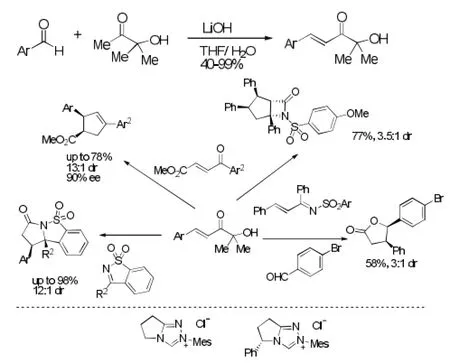
图12 [3+2]环加成反应例2Fig12 Example 2 of [3+2] cycloaddition reaction
最近,Scheidt[21]实现了N-杂环卡宾和Lewis酸共催化的高立体选择性的成环反应,拓展了N-杂环卡宾的应用范围。该反应中,N-杂环卡宾作为Lewis碱和Lewis酸共同活化底物,由于N-杂环卡宾能与后过渡金属能较牢固的结合,因此,笔者采用前过渡金属的Lewis酸。通过该方法可以制备之前无法得到的取代环戊烯,有极好的对映体选择性和非对映体选择性。最优条件下,只生成cis式的产物,产率为82%,ee值高达99%。见图13。
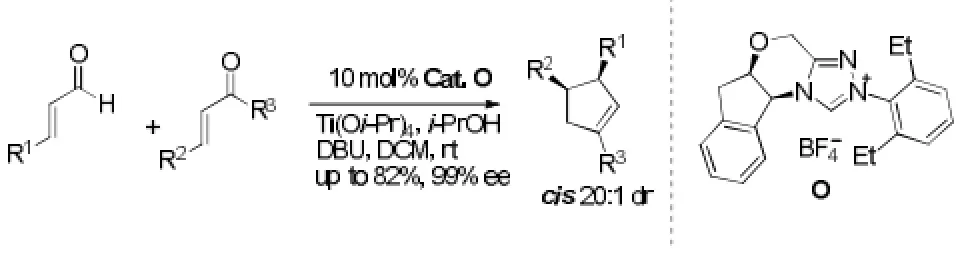
图13 [3+2]环加成反应例3Fig13 Example 3 of [3+2] cycloaddition reaction
3.3 [2+2]环加成
Ye 在N-杂环卡宾催化[2+2]环加成,合成四元环状化合物方面也同样有许多杰出的工作。2008年,Ye[22]首次报道了手性N-杂环卡宾催化的乙烯酮和N-Boc亚胺的Staudinger反应,生成N-Boc内酰胺,有很好的产率和非对映体选择性,ee值高达99%。该产物能方便的脱保护,得到内酰胺;或者通过LiAlH4还原开环得到γ-氨基醇,ee值保持不变。见图14。

图14 [2+2]环加成反应例1Fig14 Example 1 of [2+2] cycloaddition reaction
随后,Ye[23]又报道了乙烯酮和三氟甲基活化的酮的[2+2]环加成反应,高产率得到β-内酯,具有优秀的对映异构和非对映异构选择性。以手性N-杂环Q为催化剂,碳酸铯为碱,甲苯为溶剂,−40 ℃条件下,最佳达到99%的产率,96%的dr值和99%的ee值。见图15。
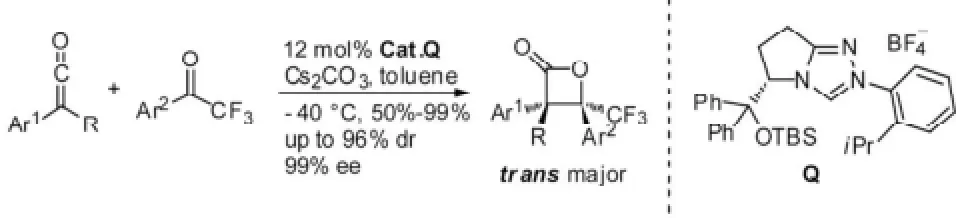
图15 [2+2]环加成反应例2Fig15 Example 2 of [2+2] cycloaddition reaction
Ye[24]还将二氮烯二羧酸酯作为亲偶极子,与乙烯酮成环,从中等到很好的收率和ee值得到氮杂-β-内酰胺。同时,对照Fu[25]和他们自己的工作[14],笔者加以研究发现,二氮烯氮上取代基的不同对控制反应类型起到非常重要的作用。羧酸酯基取代的易发生[2+2]环加成反应,而苯甲酰基取代的易发生[4+2]环加成反应,这主要是由羰基的性质决定的。见图16。
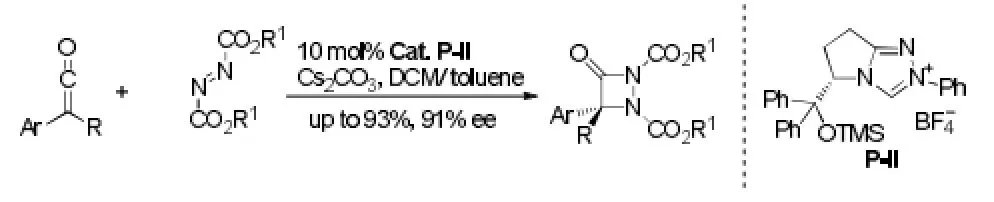
图16 [2+2]环加成反应例3Fig16 Example 3 of [2+2] cycloaddition reaction
最近,Ye[26]报道了手性NHC催化的乙烯酮和亚硝基化合物的[2+2]环加成反应,从中等到很好的收率和对映体选择性得到氧杂-β-内酰胺,产物在Zn和醋酸条件下,几乎定量的转化成具有重要合成意义的、有光学活性的α-羟基酸衍生物,且不影响ee值。见图17。
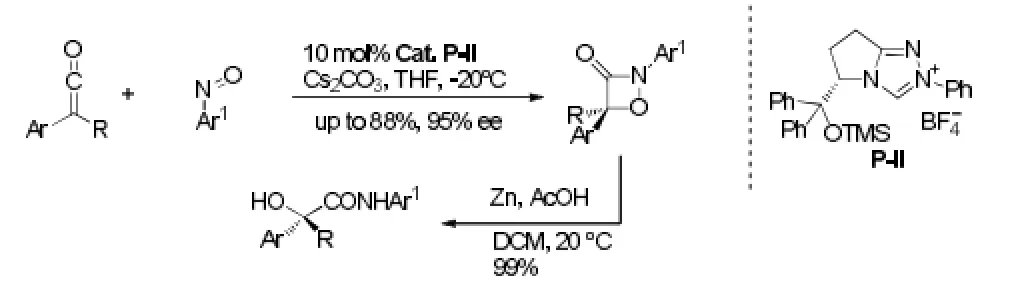
图17 [2+2]环加成反应例4Fig17 Example 4 of [2+2] cycloaddition reaction
3.4 [3+3]环加成
N-杂环卡宾催化的[3+3]环加成反应相对较少。2007年,Scheidt等[27]报道了首例NHC催化α,β-不饱和醛与偶氮甲碱亚胺的[3+3]环加成反应,立体专一性的生成双杂环化合物,有中等到很好的产率。该反应的挑战性在于体系中同时存在两个亲电底物,如何能实现NHC选择性的与α,β-不饱和醛形成加成产物。如果催化剂与亚胺发生不可逆加成,该反应则无法实现,而作者很幸运的找到了合适的亲电底物,并能得到单一的非对映体。笔者研究发现,这种高选择性是由于卡宾-醛加成物与亚胺之间形成了一个氢键,催化剂的结构迫使卡宾-醛加成物是一种舒展的立体空间,从而只能从远离偶氮甲碱环上苯基取代基的一侧进行亲核进攻。见图18。
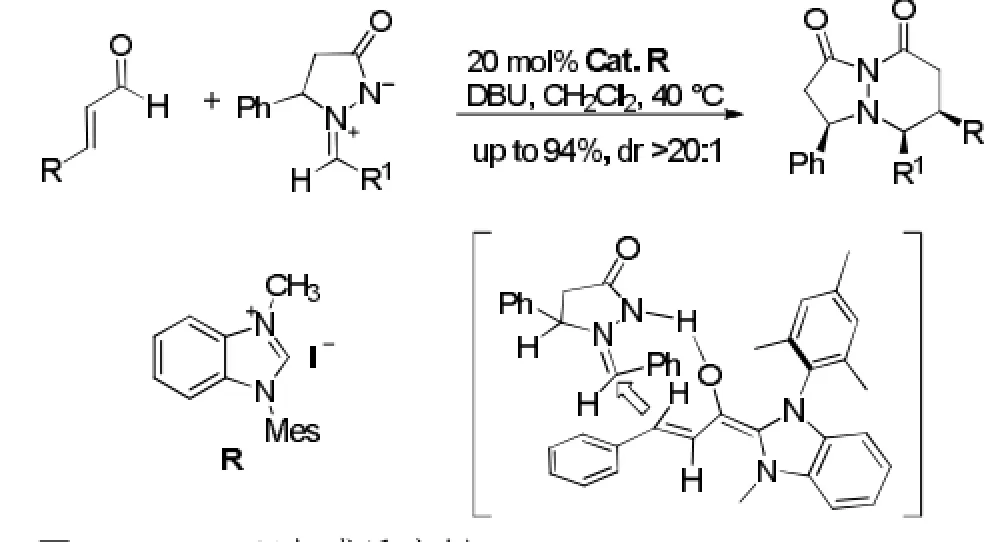
图18 [3+3]环加成反应例1Fig18 Example 1 of [3+3] cycloaddition reaction
4 串联反应
串联反应是一种高效的合成策略,可将较简单的原料通过较短的步骤转化出复杂的分子,且中间体不需要分离,简化了操作步骤。因此,将NHC催化的常见反应与其他反应类型相结合,应用于有机合成中,高效的得到环状化合物同样有很高的研究价值。
2007年,Bode等[28]报道了一种手性NHC催化环戊烯的合成方法,该反应为醛和烯酮的不对称分子间交叉安息香缩合反应和氧杂Cope重排的串联反应。在最佳反应条件下,得到的cis-环戊烯,有98%以上的ee值,产率最高达到93%;而trans-环戊烯的对映体选择性相对较差。见图19。
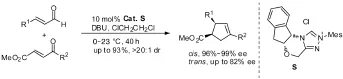
图19 串联反应例1Fig19 Example 1 of tandem Reaction
Gravel等[29]通过分子间Stetter反应与Michael加成反应的串联反应,得到一系列取代的二氢化茚产物,有较好的产率和非对映体选择性。该方法是由NHC催化醛与亲电底物的Stetter反应,产生的烯醇化物与自身的另一个缺电子基团发生Michael加成,得到环状化合物。见图20。该方法可以扩展用于多环吡咯化合物的合成,吡咯杂环是氟康唑、乙酰胺吡咯烷酮等多种药物的关键骨架结构。
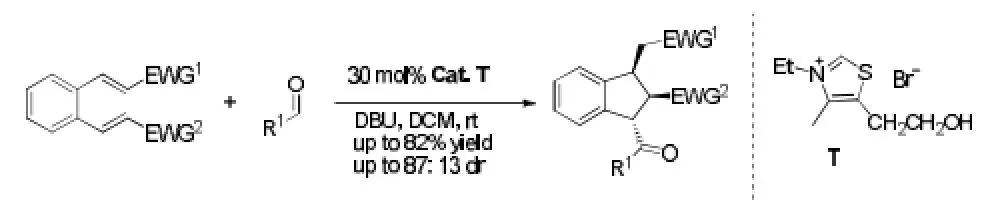
图20 串联反应例2Fig20 Example 2 of tandem Reaction
Ye等[30]在Gravel的基础上,发展出NHC催化的邻苯二醛与Michael受体的Stetter-Aldol串联反应,主产物为trans-4-羟基四氢萘酮,产率最高为72%,是合成某些海洋天然产物的重要中间体,弥补了其他合成方法的不足。而如果分步进行Stetter反应和Aldol缩合,则得到较多cis-4-羟基四氢萘酮。见图21。
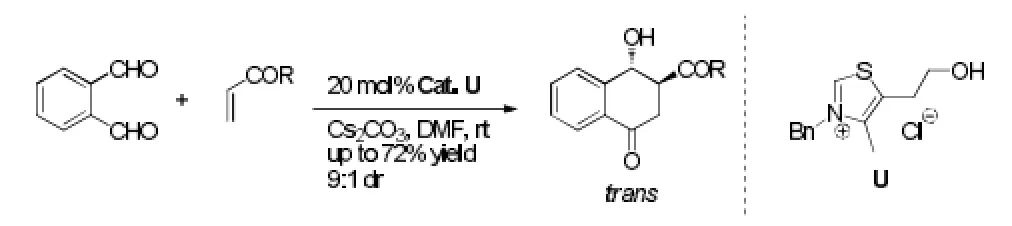
图21 串联反应例3Fig21 Example 3 of tandem Reaction
螺吲哚酮是一类结构有趣又非常重要的化合物,它广泛的存在于复杂天然产物和药物中,具有显著的生物活性。最近杜鼎课题组[31]报道了NHC催化的两分子炔醛与吲哚酮的三组分多米诺反应,以很好的收率和非对映体选择性得到含有三个相邻手性中心的螺吲哚酮衍生物。该反应经历了Breslow中间体的氧化还原反应,与吲哚酮的共轭加成,氢迁移以及与另一分子炔醛的Aldol缩合,最后通过内酯化成环。使用该咪唑类催化剂可以较好的控制Knoevenagel副产物的生成,而改用其他常用NHC催化剂将得到大量的Knoevenagel副产物。见图22。
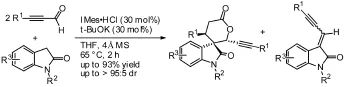
图22 串联反应例3Fig22 Example 3 of tandem Reaction
5 结论
在N-杂环卡宾催化的各种成环方法中,有的方法在区域选择性和立体选择性方面有了较大突破;有的方法产率高达90%以上;有的方法试剂温和常见,操作简便,反应时间短。N-杂环卡宾催化的各种成环方法为有机合成方法学中的高效成环,包括各种杂环和稠合环的合成提供了新的途径和策略,也为重要的医药中间体或活性化合物的合成提供了更多选择。
REFERENCES
[1] ARDUENGO III A J, HARLOW R L, KLINE M. A stable crystalline carbene [J]. J Am Chem Soc, 1991, 113(1): 361-363.
[2] BURGESS J. Transition-Metals in diagnosis and therapy [J]. Transition Met Chem, 1993, 18(4): 439-448.
[3] TAKEDA S, ABE H, TAKEUCHI Y, HARAYAMA T. Intramolecular biaryl coupling reaction of benzyl benzoate and phenyl benzoate derivatives, and its application to the formal synthesis of (-)steganone [J]. Tetrahedron, 2007, 63(2): 396-408.
[4] ENDERS D, BREUER K, RUNSINK J, et al. The first asymmetric intramolecular Stetter reaction. preliminary communication [J]. Helvetica Chimica Acta, 1996, 79(7): 1899-1902.
[5] KERR M S, ALANIZ J R, ROVIS T. A highly enantioselective catalytic intramolecular stetter reaction [J]. J Am Chem Soc, 2002, 124 (35): 10298-10299.
[6] MATSUMOTO Y, TOMIOKA K. C2 symmetric chiral N-heterocyclic carbene catalyst for asymmetric intramolecular Stetter reaction [J]. Tetrahedron Let, 2006, 47(33): 5843-5846.
[7] REYNOLDS N T, ROVIS T. The effect of pre-existing stereocenters in the intramolecular asymmetric stetter reaction [J]. Tetrahedron, 2005, 61(26,27): 6368-6378.
[8] NAKAMURA T, HARA O, TAMURA T, et al. A facile synthesis of chroman-4-ones and 2,3-dihydroquinolin-4-ones with quaternary carbon using intramolecular Stetter reaction catalyzed by thiazolium salt [J]. Synlett, 2005, (1): 155-157.
[9] LIU Q, ROVIS T. Asymmetric synthesis of hydrobenzofuranones via desymmetrization of cyclohexadienones using the intramolecular Stetter reaction [J]. J Am Chem Soc, 2006, 128(8): 2552-2553.
[10] JIA M Q, YOU S L. Desymmetrization of cyclohexadienones via D-camphor-derived triazolium salt catalyzed intramolecular Stetter reaction [J]. Chem Comm, 2012, 48: 6363-6365.
[11] FISCHER C, SMITH S W, POWELL D A, et al. Umpolung of michael acceptors catalyzed by N-heterocyclic carbenes [J]. J Am Chem Soc, 2006, 128(5): 1472-1473.
[12] RAVEENDRAN A E, PAUL R R, SURESH E, et al. Nucleophilic heterocyclic carbene as a novel catalyst for cyclopropanation of cyano acrylates [J]. Org Biomol Chem, 2010, 8(4): 901-905.
[13] ZHANG Y R, LV H, ZHOU D, et al. Chiral N-heterocyclic carbene-catalyzed formal [4+2] cycloaddition of ketenes with enones: highly enantioselective synthesis of trans- and cis-δ-lactones [J]. Chemistry, 2008, 14(28): 8473-8476.
[14] HUANG X L, HE L, SHAO P L, et al. [4+2] Cycloaddition of ketenes with N-benzoyldiazenes catalyzed by N-heterocyclic carbenes [J]. Angew Chem Int Ed Engl, 2009, 48(1): 192-195.
[15] LV H, CHEN X Y, SUN L H, et al. Enantioselective synthesis of indole-fused dihydropyranones via catalytic cycloaddition of ketenes and 3-alkylenyloxindoles [J]. J Org Chem, 2010, 75(20): 6973-6976.
[16] JIAN T Y, SHAO P L, YE S. Enantioselective [4+2] cycloaddition of ketenes and 1-azadienes catalyzed by N-heterocyclic carbenes [J]. Chem Commun, 2011, 47(8): 2381-2383.
[17] RYAN S J, CANDISH L, LUPTON D W. N-heterocyclic carbene-catalyzed generation of α,β-unsaturated acyl imidazoliums: synthesis of dihydropyranones by their reaction with enolates [J]. J Am Chem Soc, 2009, 131(40): 14176-14177.
[18] YANG L, WANG F, CHUA P J, et al. N-heterocyclic carbene (NHC)-catalyzed highly diastereo- and enantioselective Oxo-Diels-Alder reactions for synthesis of fused pyrano[2,3-b]indoles [J]. Org. Lett. 2012, 14(11): 2894-2897.
[19] CHAN A, SCHEIDT K A. Direct amination of homoenolates catalyzed by N-heterocyclic carbenes [J]. J Am Chem Soc, 2008, 130(9): 2740-2741.
[20] CHIANG P C, ROMMEL M, BODE J W. α’-Hydroxyenones as mechanistic probes and scope-expanding surrogates for α,β-unsaturated aldehydes in N-heterocyclic carbene-catalyzed reactions [J]. J Am Chem Soc, 2009, 131(24): 8714-8718.
[21] CARDINAL-DAVID B, RAUP D E A, SCHEIDT K A. Cooperative N-heterocyclic carbene/lewis acid catalysis for highly stereoselective annulation reactions with homoenolates [J]. J Am Chem Soc, 2010, 132(15): 5345-5347.
[22] ZHANG Y R, HE L, WU X, et al. Chiral N-heterocyclic carbene catalyzed staudinger reaction of ketenes with imines: highly enantioselective synthesis of N-boc β-lactams [J]. Org Lett, 2008, 10 (2): 277-280.
[23] WANG X N, SHAO P L, LV H, et al. Enantioselective synthesis of β-trifluoromethyl-β-lactones via NHC-catalyzed ketene−ketone cycloaddition reactions [J]. Org Lett, 2009, 11 (18): 4029-4031.
[24] HUANG X L, CHEN X Y, YE S. Enantioselective synthesis of aza-β-lactams via NHC-catalyzed [2+2] cycloaddition of ketenes with diazenedicarboxylates [J]. J Org Chem, 2009, 74 (19): 7585-7587.
[25] BERLIN J M, FU G C. Enantioselective nucleophilic catalysis: the synthesis of aza-β-lactams through [2+2] cycloadditions of ketenes with azo compounds [J]. Angew Chem Int Ed Enql, 2008, 47(37): 7048-7050.
[26] WANG T, HUANG X L, YE S. Enantioselective formal [2+2] cycloaddition of ketenes with nitroso compounds catalyzed by N-heterocyclic carbenes [J]. Org Biomol Chem, 2010, 8(21): 5007-5011.
[27] CHAN A, SCHEIDT K A. Highly stereoselective formal [3+3] cycloaddition of enals and azomethine imines catalyzed by N-heterocyclic carbenes [J]. J Am Chem Soc, 2007, 129(17): 5334-5335.
[28] CHIANG P C, KAEOBAMRUNG J, BODE J W. Enantioselective, cyclopentene-forming annulations via NHC-catalyzed benzoin−oxy-Cope reactions [J]. J Am Chem Soc, 2007, 129 (12): 3520-3521.
[29] SANCHEZ-LARIOS E, GRAVEL M. Diastereoselective synthesis of indanes via a domino Stetter-Michael reaction [J]. J Org Chem, 2009, 74(19): 7536-7539.
[30] SUN F G, HUANG X L, YE S. Diastereoselective synthesis of 4-hydroxytetralones via a cascade Stetter-Aldol reaction catalyzed by N-heterocyclic carbenes [J]. J Org Chem, 2010, 75(1): 273-276.
[31] DU D, HU Z, JIN J. et al. N-heterocyclic carbene-catalyzed three-component domino reaction of alkynyl aldehydes with oxindoles [J]. Org Lett, 2012, 14(5): 1274-1277.
Process in N-Heterocyclic Carbene-Catalyzed Annulation Reactions
NI Jiating, DONG Jing, HU Wenjun, DU Wenting*(Department of Pharmacy, Zhejiang Medical College, Hangzhou 310053, China)
OBJECTIVE To introduce the process in N-heterocyclic carbene-catalyzed annulation reactions. METHODS To review both domestic and international published reports regarding N-heterocyclic carbene-catalyzed annulation reactions. RESULTS To make a list of N-heterocyclic carbene-catalyzed annulation reactions of intramolecular Stetter reaction, nucleophilic substitution reaction, cycloaddition and tandem reaction and to analyze N-heterocyclic carbene-catalyzed [4+2], [3+2], [2+2] and [3+3] cycloaddition specially. CONCLUSION The development of the N-heterocyclic carbene-catalyzed annulation reactions provides us new ways and strategies for efficient annulation.
N-heterocyclic carbene (NHC); annulation reactions; catalyze
R573
A
1007-7693(2013)05-0558-07
2012-10-25
国家自然科学基金项目(21002026);2012年浙江省大学生科技创新活动计划(新苗人才计划2012R434006);浙江医学高等专科学校博士启动基金项目(No.2011B01)
倪佳婷,女,学生 Tel: 13735806569 E-mail: 445728550@qq.com*
杜文婷,女,博士,副教授 Tel: 13588062633 E-mail: ddwwtt@163.com
猜你喜欢
高等学校化学学报(2024年2期)2024-03-06 06:31:12
科学技术与工程(2020年34期)2021-01-08 05:43:32
世界农药(2019年4期)2019-12-30 06:25:08
中成药(2017年9期)2017-12-19 13:34:31
合成化学(2015年2期)2016-01-17 09:03:25
合成化学(2015年9期)2016-01-17 08:57:21
时尚北京(2015年1期)2015-01-30 00:00:35
无机化学学报(2014年6期)2014-02-28 17:31:59
无机化学学报(2014年1期)2014-02-28 17:30:01
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
