
CoPt纳米空心球甲醇电催化氧化和原位电化学傅里叶变换红外光谱研究
2012-11-30 10:48周新文甘亚利孙世刚
物理化学学报 2012年9期
周新文 甘亚利 孙世刚
(1三峡大学化学与生命科学学院化学系,湖北宜昌443002; 2厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门361005)
CoPt纳米空心球甲醇电催化氧化和原位电化学傅里叶变换红外光谱研究
周新文1,*甘亚利1孙世刚2,*
(1三峡大学化学与生命科学学院化学系,湖北宜昌443002;2厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门361005)
采用化学还原和电位置换法制备了CoPt纳米空心球,该催化剂对甲醇氧化表现出较好的电催化活性.透射电镜(TEM)、能量散射光谱(EDS)和电化学循环伏安实验结果表明,在0.1 mol·L-1H2SO4+0.1 mol·L-1CH3OH中进行测试时,CoPt纳米空心球发生了去合金化过程,催化剂表面Co元素溶解,形成了富Pt表面,表现出更好的电催化活性,同时表现出较好的结构稳定性.采用原位电化学红外光谱在分子水平研究了CoPt纳米空心球上甲醇氧化过程,发现甲醇在CoPt纳米空心球氧化中间产物主要为CO,且CO表现出异常红外效应,与CO为探针分子在CoPt纳米空心球上得到的红外光谱结果一致.研究结果表明,去合金化方法是一种有效调节催化剂表面组成和性能的手段,原位电化学红外光谱是潜在的原位研究有机小分子氧化机理的方法,在燃料电池中将得到广泛的应用.
CoPt纳米空心球;去合金化;甲醇氧化;原位电化学红外光谱;燃料电池
1 引言
Pt基纳米催化剂对甲醇、乙醇、甲酸等有机小分子的氧化有很好的催化性能,同时对于氧还原也表现出较好的催化活性.但Pt基催化剂存在价格昂贵、催化活性和稳定性有待进一步提高等不足.因此,Pt基燃料电池纳米催化剂的研究一直是燃料电池研究的热点问题之一.如何在减少贵金属Pt的同时,提高其催化性能是燃料电池Pt基催化剂面临的一大难题.1,2
目前Pt基燃料电池纳米催化剂的研究主要集中在以下几个方面.(1)PtM合金纳米催化剂:合成Pt基合金纳米催化剂来减少贵金属Pt的用量,同时利用金属M来提高其稳定性.3,4(2)高指数晶面Pt基纳米材料的研究:高指数晶面的催化活性要高于常规晶面,采用化学法或电化学法合成具有高指数晶面的Pt纳米材料来提高单位质量Pt的催化性能也是目前的研究热点之一.5,6(3)特殊结构Pt基纳米催化剂的合成:空心结构和核壳结构Pt基催化剂主要在于保持或提高其电催化活性的同时,减少贵金属Pt的用量.7,8(4)非Pt催化剂的研究:制约燃料电池商业化的一个重要问题是昂贵的催化剂,除了前面提到一些改进Pt基催化剂的方法之外,人们对非Pt基催化剂在直接醇类燃料电池中的研究也一直在进行.Kua和Goddard9利用非定域密度结构理论计算方法研究了VIII族过渡金属对甲醇氧化的电催化能力,结果发现,单金属锇(Os)对甲醇氧化具有较高的催化活性,本身抗一氧化碳毒化能力强于铂,有希望成为单组分催化剂.其余一些非Pt催化剂也有报道,但和Pt基催化剂相比,还没有很大的突破.10
(5)高效碳载体的研究:载体性质对催化剂的性能有较大的影响,目前主要采用碳材料作为载体.研究开发具有好的导电性、较大的表面积、合理的孔结构和优异的抗腐蚀性能等特点的碳材料也是提高催化剂性能的有效途径之一.11空心结构纳米材料由于具有密度小、比表面积大、经济等优点引起人们的广泛关注,在直接甲醇燃料电池中,Pt基空心结构纳米材料也成为改善Pt纳米粒子催化性能的有效途径之一.空心结构Pt、12Pd、13CoPt、14PtNi、15PtSe、16PtRu、17PtPb、18PtCu19等纳米催化剂均有报道.前期我们20采用化学还原和电位置换法制备了CoPt纳米空心球,该催化剂表现出比商业Pt/C催化剂更好的性能.
本文在前期工作基础上,采用电化学循环伏安法结合透射电子显微镜和能量散射光谱,进一步深入研究CoPt纳米空心球表现出较好催化活性的原因.同时采用原位电化学红外光谱研究了CoPt纳米空心球上甲醇电化学氧化过程.
2 实验
2.1 试剂和仪器
氯化钴、聚乙烯吡咯烷酮、硼氢化钠、氯铂酸钾、无水甲醇等均为分析纯.5%Nafion溶液为Alfa Aesar公司产品.电解液用硫酸为优级纯.所有溶液均以去离子水配置.
电化学实验采用上海辰华仪器公司生产的CHI 631C电化学分析仪.原位电化学红外光谱采用Nexus 870傅里叶变换红外光谱仪(美国Thermo Nicolet公司),配备有液氮冷却的MCT-A型检测器和EverGloTM红外光源,PRAC-263型恒电位仪(美国EG&G公司).透射电镜(TEM)和能量散射光谱(EDS)在Tecnai F30电子显微镜上进行(荷兰Philip-FEI公司),加速电压300 kV.
2.2 CoPt纳米空心球的合成及电极的制备
CoPt纳米空心球采用化学还原和电位置换法制备.208.5 mg氯化钴和200 mg聚乙烯吡咯烷酮溶于60 mL水,通入高纯氮气15 min,磁力搅拌,然后加入新制的硼氢化钠(NaBH4)溶液(200 mg溶于50 mL水),滴加完后,迅速加入氯铂酸钾(K2PtCl6· 6H2O)溶液(32.8 mg溶于40 mL水),继续反应1-2 h,整个实验温度控制在30°C.离心分离,洗涤,得到的产物重新分散于超纯水中备用.在进行电化学和红外实验时,移取一定量体积的纳米粒子于玻碳电极上,滴加Nafion溶液,制备的电极记为CoPt/GC.
2.3 循环伏安实验和原位电化学红外光谱实验
电化学循环伏安实验在三电极体系电解池中进行,辅助电极为铂黑电极,参比电极为饱和甘汞电极(SCE),电解质为0.1 mol·L-1H2SO4和0.1 mol· L-1H2SO4+0.1 mol·L-1CH3OH溶液.电化学实验前通高纯氮气15 min除去溶液中氧气.
原位电化学红外光谱实验过程中光学台内部由除去水气和CO2的洁净空气吹扫.原位红外光谱实验中的电极电位由PRAC-263型恒电位仪控制,并通过自行设计的接口和软件使之与红外数据的采集保持同步.原位电化学红外光谱实验采用如下步骤:将制得的电极先在0.1 mol·L-1H2SO4中清洗以活化电极表面,然后将清洗得到的电极转移到装有0.1 mol·L-1H2SO4和0.1 mol·L-1CH3OH溶液的红外电解池中进行红外光谱实验.首先将电位阶跃至0.90 V,停留5 s,将电极表面吸附物种氧化,再将电位阶跃至-0.15 V,停留5 s,使电极附近的溶液重新达到平衡,采集参考电位下的单光束光谱R(ER),然后设定研究电位的变量(ΔES)为50 mV,从-0.15 V开始逐步升高研究电位ES至0.8 V,采集一系列研究电位下的单光束光谱R(ES),最后按下式进行差减得到结果光谱.

3 结果与讨论
3.1 CoPt纳米空心球的去合金化
在前期工作20当中,我们将空心结构CoPt纳米催化剂对甲醇氧化表现出较好催化活性归功于其双功能机理和特殊的空心结构.进一步研究表明,除了这两个因素之外,还有一个很重要的原因,即去合金化过程.去合金化是一种调节催化剂表面组成和电子效应的有效手段.可以通过化学法或电化学法将合金中性质较活拨的金属溶解后得到表面富含另一种金属的催化剂,能够进一步提高电催化性能.21图1为CoPt电极在甲醇氧化前后的循环伏安(CV)曲线,扫描速率100 mV·s-1.图1a为CoPt/GC电极在0.1 mol·L-1H2SO4中的CV图.CoPt纳米空心球在0.1 mol·L-1H2SO4+0.1 mol·L-1CH3OH中扫描至稳定后,再将电极转移至0.1 mol·L-1H2SO4中得到的CV图如图1b所示.通过比较可以发现,在-0.25-0.20 V区间氢的吸脱附特征峰明显增强,正向扫描中0.65 V左右Pt的氧化峰和回扫时0.45 V左右Pt氧化物的还原峰明显增大,说明Co元素逐渐溶出,在甲醇氧化过程中发生了去合金化过程,露出更多Pt的活性位,在循环伏安图中表现出更明显的Pt的特征峰.因此对甲醇氧化表现出更高的电催化活性.

图1 0.1 mol·L-1H2SO4溶液中CoPt/GC电极在甲醇氧化实验前后的循环伏安曲线Fig.1 Cyclic voltammograms of CoPt/GC electrode in 0.1 mol·L-1H2SO4solution before and after the CH3OH oxidation
通过上面的分析我们知道,CoP纳米催化剂在酸性体系中,在CH3OH氧化前后循环伏安曲线发生较明显的变化,说明表层的Co元素发生了去合金化过程.那么,Co元素的溶解会不会破坏CoPt纳米催化剂特殊的空心结构呢?我们收集了电化学测试后的CoPt纳米催化剂样品并对它们做了进一步表征.图2为甲醇氧化实验前、后CoPt纳米空心球的TEM和EDS图.图2a表明CoPt纳米催化剂为空心链状结构,EDS结果表明平均组成为Co20Pt80.电化学实验之后CoPt纳米催化剂保持较好的一维链状空心结构,表明Co元素的溶解并没有破坏其结构,部分纳米球只是简单地堆积在一起,没有发生团聚.从EDS图(图2d)中可以看出,更多Pt元素对应的谱峰增强,电化学实验之后CoPt纳米催化剂平均组成为Co13Pt87,说明表层Co元素的确发生了溶解,与前面的分析结果一致.
3.2 CoPt纳米空心球甲醇氧化的原位FTIR光谱
采用原位电化学红外光谱研究了甲醇在CoPt纳米催化剂上的电催化氧化行为.由于随着研究电位改变,背景光谱变化较大,因此我们在不同电位范围内研究了结果光谱.图3给出的是研究电位在-0.15-0.45 V之间的结果光谱,图4给出的是研究电位在0.50-0.80 V之间的结果光谱.可以看出,在2000-2100 cm-1范围内,随着研究电位的升高,在2055 cm-1附近逐渐出现了一正向谱峰,该谱峰为CH3OH毒性中间体产物线型CO(COL)的红外吸收峰.根据差谱定义,我们知道,正向谱峰对应于研究电位下红外反射率的提高.由图中可以看出,在一定研究电位内,随着电位升高,谱峰的峰位逐渐向高波数移动,即吸附于CoPt/GC电极上的线型CO具有典型的电化学Stark效应,得到的Stark系数为55.9 cm-1·V-1(如图5所示),大于本体Pt电极上吸附态CO的Stark系数.20
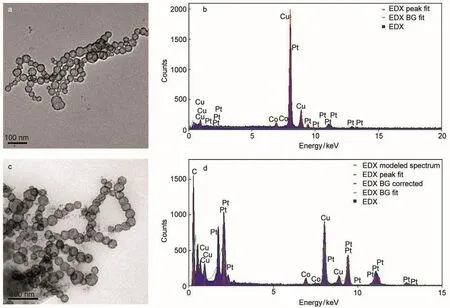
图2 甲醇氧化前(a,b)后(c,d)CoPt纳米催化剂透射电镜(TEM)照片(a,c)和能量散射光谱(EDS)图(b,d)Fig.2 Transmission electron microscopy(TEM)images(a,c)and energy dispersive spectrometer(EDS)patterns(b,d)of CoPt nanocatalyst obtained before(a,b)and after(c,d)the CH3OH oxidation

图3 CoPt/GC电极在0.1 mol·L-1H2SO4+0.1 mol·L-1 CH3OH溶液中原位FTIR光谱Fig.3 In situ FTIR spectra recorded on CoPt/GC electrode in 0.1 mol·L-1H2SO4+0.1 mol·L-1CH3OH solutionΔR/R:relative change in the electrode reflectivity; studying potential(ES):-0.15-0.45 V

图4 CoPt/GC电极在0.1 mol·L-1H2SO4+0.1 mol·L-1 CH3OH溶液中原位FTIR光谱Fig.4 In situ FTIR spectra recorded on CoPt/GC electrode in 0.1 mol·L-1H2SO4+0.1 mol·L-1CH3OH solution ES:0.50-0.80 V

图5 图3中线型CO(COL)谱峰位置随研究电位ES的变化曲线Fig.5 Plot of linearly bonded CO(COL)band center versus studying potential ESmeasured from Fig.3
对图3中COL谱峰的面积进行积分得到COL谱峰的强度随电位变化如图6所示.可以看到,从-0.05 V开始,吸附在CoPt/GC电极上COL的量迅速增加,在0.20 V左右达到最大值.当研究电位继续升高时, COL的量迅速减少.图3可以观察到当电位高于0.45 V时,COL的红外吸收峰基本消失.说明在较低的电位区间,CoPt/GC电极表面主要为甲醇解离产物CO所覆盖,致使甲醇的氧化不能进行.当ES≥0.20 V时,在2345 cm-1处出现一生成物种的负向谱峰,为CO2不对称伸缩振动的红外吸收峰,该谱峰没有Stark效应,表明CO2存在于薄层溶液中.CO2谱峰积分强度(谱峰面积)随电位的变化如图7所示.当电位低于0.20 V时,基本上没有CO2产生,说明在0.20 V以下,毒性中间体CO能稳定地存在于电极表面,与图6中CO强度随电位变化结果一致.当电位高于0.25 V时,CO2的量迅速增加,并于0.50 V左右达到最大,这是由于表面吸附态的CO在电位高于0.25 V时被逐渐氧化,使得表面活性位增多,甲醇开始氧化产生大量CO2引起的.
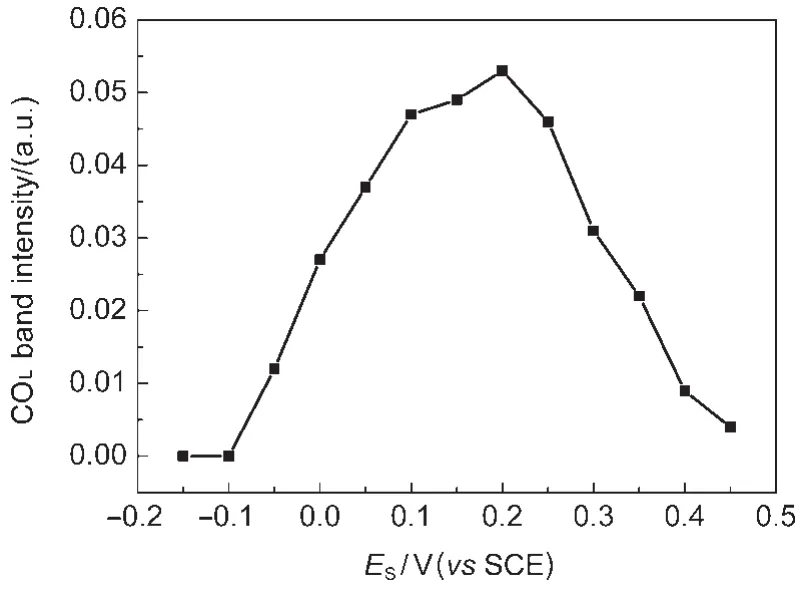
图6 图3中COL谱峰强度随样品电位ES的变化曲线Fig.6 Variation of integrated band intensity of COLversus studying potential ESmeasured from Fig.3

图7 图3和图4中CO2谱峰强度随研究电位ES的变化曲线Fig.7 Variation of integrated band intensity of CO2versus studying potential ESmeasured from Fig.3 and Fig.4
从图3中可以看出,甲醇氧化中解离产生的CO给出单极的正向谱峰,表现为异常红外效应.与文献18中CO吸附在空心结构CoPt/GC电极上得到的异常红外效应结果一致.需要指出的是,陈卫等22采用原位电化学红外光谱发现,甲醇氧化解离产生的CO吸附在团聚态Pt纳米粒子上给出左高右低的双极谱峰.Li等23采用原位电化学显微红外光谱发现甲醇解离产生的CO吸附在不同结构Pt纳米粒子薄膜上时,随着氧化电位升高,先给出左高右低的双极谱峰,再变成单极向下的红外吸收峰后转变为左低右高的双极峰.Tkach等24在不同Pt基催化剂上同样观察到毒性中间体CO给出不同的谱峰形状.说明在甲醇氧化过程中,毒性中间体CO的谱峰形状与纳米催化剂的组成、尺寸和聚集状态等密切相关.
当电位高于0.35 V之后,红外谱图发生了较大的变化,为了更清楚地分析谱图,0.50-0.80 V电位范围内的结果光谱如图4所示.1065 cm-1附近的谱峰归属为甲醇中C―O键的伸缩振动吸收峰,该峰方向朝上对应于薄层中甲醇的消耗.1046和1185 cm-1附近的谱峰为HSO4-的吸收峰,该峰谱峰朝下,表示有HSO4-的生成,这是由于甲醇在氧化过程中会产生H+,使得溶液中HSO4-的量增加引起的.另外,陈卫22和Li23等在用纯Pt做催化剂催化氧化甲醇时,原位FTIR中在1643 cm-1附近均能观察到一个很强的正向谱峰,归属于水的HOH弯曲振动吸收峰,意味着薄层中水的消耗.但这里我们并没有很明显地观察到这个谱峰,可能是由于Co元素的加入改变了Pt的电子结构,使得在甲醇氧化过程中水的消耗减少引起的.
4 结论
采用电化学循环伏安法、透射电子显微镜、能量散射光谱和原位电化学红外光谱方法研究了CoPt纳米空心球上甲醇的电化学氧化过程.结果表明,在电化学实验过程中,CoPt纳米空心球发生了去合金化过程,表层的Co元素溶解,表面裸露出更多Pt的活性位,进而提高了其对甲醇的电催化氧化能力.原位红外光谱结果表明,甲醇解离吸附毒性物种CO在2055 cm-1附近的正向谱峰,表吸出异常红外效应,测得的Stark系数为55.9 cm-1·V-1.随着研究电位的升高,CO谱峰强度先增大后减小,在0.40 V左右基本完全被氧化,与此同时,CO2谱峰强度急剧增大,表明毒性中间体CO在电位高于0.20 V时逐渐发生氧化,同时甲醇在电极上逐渐开始氧化.甲醇解离产生的毒性中间体CO在空心CoPt/GC电极上表现出异常红外效应.
(1) Hou,M.;Yi,B.L.J.Electrochem.2012,18(1),1.[侯 明,衣宝廉.电化学,2012,18(1),1.]
(2) Sun,Y.B.;Zhuang,L.;Lu,J.T.;Hong,X.L.;Liu,P.F.J.Am. Chem.Soc.2007,129,15465.doi:10.1021/ja076177b
(3)Anderson,A.B.;Grantscharora,E.;Seong,S.J.Electrochem. Soc.1996,43(6),2075.
(4)Nakagawa,N.;Kaneda,Y.;Wagatsuma.M.;Tsujiguchi,T. J.Power Sources 2012,199,103.doi:10.1016/j. jpowsour.2011.10.057
(5)Tian,N.;Zhou,Z.Y.;Sun,S.G.;Ding,Y.;Wang,Z.L.Science 2007,316,762.
(6)Tian,N.;Zhou,Z.Y.;Yu,N.F.;Sun,S.G.J.Am.Chem.Soc. 2010,132,7580.doi:10.1021/ja102177r
(7) Liu,B.;Liao,S.J.;Liang,Z.X.Prog.Chem.2011,23(5), 852. [刘 宾,廖世军,梁振兴.化学进展,2011,23(5),852.]
(8)Ataee-Esfahani,H.;Nemoto,Y.;Wang,L.;Yamauchi,Y.Chem. Commun.2011,47(13),3885.doi:10.1039/c0cc05233g
(9) Kua,J.;Goddard,W.A.J.Am.Chem.Soc.1999,121,10928. doi:10.1021/ja9844074
(10) Chen,Z.W.;Higgins,D.;Yu,A.P.;Zhang,L.;Zhang,J.J. Energ.Environ.Sci.2011,4(9),3167.doi:10.1039/c0ee00558d
(11) Luo,B.M.;Yan,X.B.;Xu,S.;Xue,Q.J.Electrochim.Acta 2012,59(1),429.
(12)Liang,H.P.;Zhang,H.M.;Hu,J.S.;Guo,Y.G.;Wan,L.J.; Bai,C.L.Angew.Chem.Int.Edit.2004,43,1540.doi:10.1002/ anie.200352956
(13) Ge,J.;Xing,W.;Xue,X.;Liu,C.;Lu,T.;Liao,J.J.Phys. Chem.C 2007,111,17305.doi:10.1021/jp073666p
(14)Chen,G.;Xia,D.;Nie,Z.;Wang,Z.;Wang,L.;Zhang,L.; Zhang,J.Chem.Mater.2007,19,1840.doi:10.1021/ cm062336z
(15)Zhou,X.W.;Zhang,R.H.;Zhou,Z.Y.;Sun,S.G.J.Power Sources 2011,196,5844.doi:10.1016/j.jpowsour.2011.02.088
(16)Yan,L.L.;Jiang,Q.N.;Liu,D.Y.;Zhong,Y.;Wen,P.F.;Deng, X.C.;Zhong,Q.L.;Ren,B.;Tian,Z.Q.Acta Phys.-Chim.Sin. 2010,26(9),2337.[颜亮亮,姜庆宁,刘德宇,钟 艳,温飞鹏,邓小聪,钟起玲,任 斌,田中群.物理化学学报,2010,26
(9),2337.]doi:10.3866/PKU.WHXB20100835
(17)Minch,R.;Es-Souni,M.Chem.Commun.2011,47(22),6284. doi:10.1039/c1cc11398d
(18)Chen,D.J.;Zhou,Z.Y.;Wang.Q.;Xiang,D.M.;Tian,N.;Sun, S.G.Chem.Commun.2010,46(24),4252.doi:10.1039/ c002964e
(19)Yu,X.F.;Wang,D.S.;Peng,Q.;Li,Y.D.Chem.Commun. 2011,47(28),8094.doi:10.1039/c1cc12416a
(20) Zhou,X.W.;Chen,Q.S.;Zhou,Z.Y.;Sun,S.G.J.Nanosci. Nanotech.2009,9(4),2392.doi:10.1166/jnn.2009.SE34
(21) Strasser,P.;Koh,S.;Anniyev,T.;Greeley,J.;More,K.;Yu,C. F.;Liu,Z.C.;Kaya,S.;Nordlund,D.;Ogasawara,H.;Toney, M.F.;Nilsson,A.Nat.Chem.2010,2(6),454.doi:10.1038/ nchem.623
(22) Chen,W.;Sun,S.G.;Si,D.;Chen,S.P.Acta Phys.-Chim.Sin. 2003,19(5),441.[陈 卫,孙世刚,司 迪,陈声培.物理化学学报,2003,19(5),441.]doi:10.3866/PKU.WHXB20030513
(23) Li,J.T.;Chen,Q.S.;Sun,S.G.Electrochim.Acta 2007,52, 5725.doi:10.1016/j.electacta.2006.12.082
(24) Tkach,I.;Panchenko,A.;Kaz,T.;Gogel,V.;Friedrich,K.A.; Roduner,E.Phys.Chem.Chem.Phys.2004,6,5419.
March 7,2012;Revised:May 2,2012;Published on Web:May 3,2012.
Studies of Oxidation Processes of Methanol on Hollow CoPt Nanospheres and In situ Electrochemical Fourier Transform Infrared Spectroscopy
ZHOU Xin-Wen1,*GAN Ya-Li1SUN Shi-Gang2,*
(1Department of Chemistry,College of Chemistry and Life Science,Three Gorges University,Yichang 443002,Hubei Province,P.R. China;2State Key Laboratory of Physical Chemistry of Solid Surface,Department of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,Fujian Province,P.R.China)
Hollow CoPt nanospheres were synthesized by chemical reduction and galvanic displacement reactions.The catalyst showed good electrocatalytic activity for methanol oxidation.The results of transmission electron microscopy(TEM),energy dispersive spectromenter(EDS),and electrochemical cyclic voltammograms indicated that,in the process of electrochemical experiments carried in 0.1 mol·L-1H2SO4and 0.1 mol·L-1CH3OH,hollow CoPt nanospheres were dealloying,which induced the dissolution of elemental Co from the surface of the catalyst.After the dealloying process,more Pt active sites were exposed on the surface of the catalyst and the catalyst showed better catalytic activity,as well as enhanced structural stability.The electrooxidation of methanol on the hollow CoPt nanospheres was studied on the molecular level using in situ electrochemical Fourier transform infrared(FTIR)spectroscopy.The toxic intermediate CO observed on the CoPt nanorods displayed abnormal infrared effects(AIREs).The FTIR results were similar to those obtained in an earlier experiment on the hollow CoPt nanospheres using CO as a probe molecule.All the results suggested that the dealloying method would be a useful technique for regulating the composition and performance of the catalyst.In situ electrochemical FTIR was highlighted as a potential method for studying the oxidation processes of organic molecules.It is envisaged that these methods will be widely used in the field of fuel cell research.
Hollow CoPt nanosphere;Dealloying process;Methanol oxidation; In situ electrochemical FTIR spectroscopy;Fuel cell
10.3866/PKU.WHXB201205031
O646
∗Corresponding authors.ZHOU Xin-Wen,Email:xwzhou@ctgu.edu.cn.SUN Shi-Gang,Email:sgsun@xmu.edu.cn;Tel:+86-592-2180181. The project was supported by the National Natural Science Foundation of China(20833005).
国家自然科学基金(20833005)资助项目
猜你喜欢
石油科学通报(2021年3期)2021-10-14
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
分析科学学报(2021年3期)2021-07-14
色谱(2021年6期)2021-05-06
陶瓷学报(2020年6期)2021-01-26
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
当代化工(2020年6期)2020-08-24
科技资讯(2020年12期)2020-06-03
空天防御(2020年1期)2020-04-13
表面工程与再制造(2019年6期)2019-08-24
