
芬太尼对慢性吗啡耐受大鼠蓝斑核δ受体与β-arrestin 1的作用①
2012-11-27 06:20刘若杉孙莉
中国康复理论与实践 2012年4期
刘若杉,孙莉
吗啡作为经典的阿片类药物,是治疗中、重度疼痛最常用、最为有效的镇痛药物。但实际临床应用中,长期、大剂量使用该药常出现吗啡耐受现象,药物镇痛作用逐渐减退甚至消失,还可出现严重的痛觉过敏,明显降低疼痛治疗效果,在很大程度上限制了吗啡的临床应用。
δ受体是慢性吗啡耐受形成的重要分子基础,在此过程中其表达明显减少[1],而β-arrestin 1作为参与阿片受体脱敏与内吞的可溶性蛋白,主要介导δ受体脱敏与内吞。研究证实,小剂量芬太尼可延缓大鼠慢性吗啡耐受的出现,但其机制仍需进一步探讨[2]。本研究拟观察不同剂量芬太尼对慢性吗啡耐受大鼠蓝斑核δ受体与β-arrestin 1表达的可能影响,初步探讨其延缓慢性吗啡耐受发生的机制。
1 材料和方法
1.1 材料 成年雄性SD大鼠40只,体重(230±20)g,由维通利华实验动物技术有限公司提供;硫酸吗啡注射液(宜昌人福药业有限责任公司,批号:080513);枸橼酸芬太尼注射液(宜昌人福药业有限责任公司,批号:071106);δ受体多克隆抗体(Chemicon公司,美国);β-arrestin 1多克隆抗体(Santa Cruz Biotechnology公司,美国);RT-PCR试剂盒(Takara公司,日本);ZH-LUO/B鼠尾测痛仪(淮北正华生物仪器设备有限公司)。
1.2 实验分组与给药方法 全部实验动物置于22℃、12 h/12 h昼夜照明的环境中,适应环境3 d后开始实验。实验前对大鼠进行痛阈筛选,排除痛反应时间超过(xˉ±3SD)的大鼠。动物随机分为5组(n=8):对照组(NS组):注射生理盐水1 ml/kg 2次,间隔30 min;慢性吗啡耐受组(M组):注射吗啡10 mg/kg,30min后给予生理盐水1 ml/kg;MF1、MF2、MF3组:吗啡用药剂量同M组,给药30 min后分别给予芬太尼3 μg/kg、6 μg/kg、12 μg/kg。采用颈部皮下注射给药,每天给药2次(8∶00、16∶00),连续9 d。
1.3 热辐射甩尾法痛阈行为学测试 每天于上午注射给药后30 min测定大鼠痛阈。将大鼠置于固定器内,待其安静后将鼠尾放置于测痛仪的凹槽中,开启50 W卤素灯,照射鼠尾中后1/3处,记录从热源启动到大鼠甩尾的时间作为甩尾反应的潜伏期(TFL阈值)。分别测量3次,每次间隔5 min,取其平均值作为痛阈值。设定终止时间为12 s,以防大鼠尾部皮肤损伤。
1.4 δ受体与β-arrestin 1 mRNA表达的测定 全部实验动物于第9天测痛阈值后,快速断头处死,取蓝斑核部位脑组织。按照Trizol试剂说明书提取总RNA,测定RNA浓度,取2 μg总RNA,按照反转录试剂盒说明书进行反转录反应,制备cDNA。β-actin、δ受体、β-arrestin 1的特异性PCR引物如下:β-actin上游引物为 5′-CATCTCTTGCTCGAAGTCCA-3′, 下 游 引 物 为5′-ATCATGTTTGAGACC-TTCAACA-3′;δ受体上游引物为 5′GCATCTGGG TCTTGGCTTCA3′,下游引物为 5′GGCTGCGGTCCTTCTCCTT3′;β-arrestin 1 上游引物为 5′CGCCAACCGTGA-AATCCT3′,下游引物为5′CCATCATCCTCTTCGTCCT3′。
1.5 δ受体与β-arrestin 1蛋白表达的测定 取蓝斑核部位脑组织,以300 μl全细胞蛋白裂解液进行组织匀浆,提取组织蛋白(整个操作过程在冰浴中进行),测定蛋白浓度后,-80℃保存。取细胞总蛋白50µg,适量loading Buffer混匀,95℃水浴处理5 min,经12%SDS-PAGE电泳后,转到硝酸纤维素膜上,分别加入1∶500倍稀释的δ受体与1∶1000倍稀释的β-arrestin 1,室温孵育1 h,再加入1∶5000倍稀释的HRP标记的羊抗兔IgG,曝光、显影后分析条带灰度,校正各蛋白的表达水平,分析δ受体与β-arrestin 1的蛋白表达。
1.6 统计学分析 应用SAS 9.1.3统计软件进行统计学处理。计量资料以(±s)表示。各组间比较采用两因素方差分析(two-way ANOVA),应用Dunnett-t检验进行多重比较。
2 结果
2.1 大鼠痛阈值 给药前,各组大鼠痛阈值无显著性差异(P>0.05)。M组大鼠注射吗啡后,痛阈值较NS组明显升高(P<0.01),随着用药次数增加,痛阈值逐渐降低,第9天痛阈值降至给药前水平。MF2组与MF3组大鼠痛阈值的降低较M组缓慢,第7、9天MF2组与MF3组痛阈值均高于M组(P<0.05);MF1组大鼠注射吗啡后痛阈值无改变(P>0.05)。见表1。
2.2 δ受体表达 与NS组比较,M组δ受体mRNA及其蛋白表达明显减少(P<0.01);与M组比较,MF1、MF2和MF3组δ受体mRNA及其蛋白表达无显著性差异(P>0.05)。见图1、图2。

表1 各组大鼠给药前后痛阈值的比较(s)


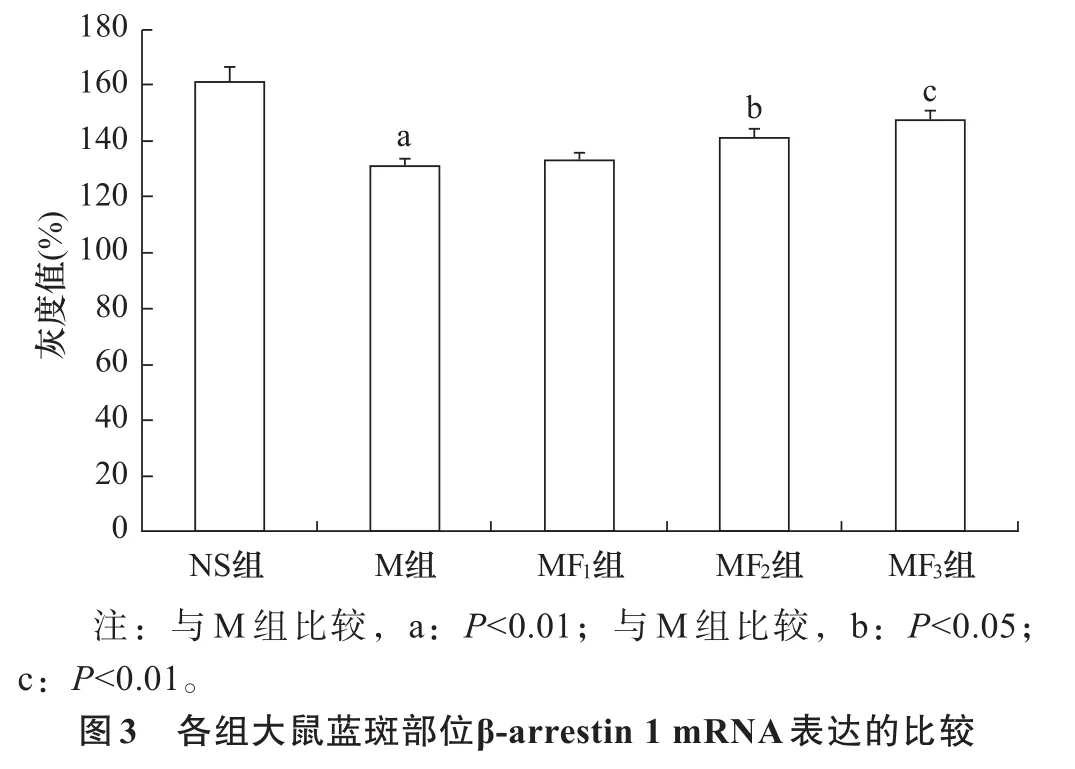
2.3 β-arrestin 1表达 与NS组比较,M组β-arrestin 1 mRNA及其蛋白表达明显减少(P<0.01);与M组相比,MF2和MF3组β-arrestin 1 mRNA及其蛋白表达呈剂量依赖性增加(P<0.05),而MF1组则无显著性差异(P>0.05)。见图3、图4。

3 讨论
建立慢性吗啡耐受动物模型的方法有多种,通过不同给药剂量及途径均可完成。衡量模型建立是否成功的主要标准为实验动物甩尾潜伏期在药物使用后延长,并在给药结束后恢复至给药前水平[3]。实验中,我们应用连续颈部皮下注射吗啡的方法来建立大鼠慢性吗啡耐受模型,将大鼠热辐射甩尾潜伏期作为痛阈指标,评价大鼠慢性吗啡耐受的形成。本研究结果显示,与NS组相比,M组大鼠第1天皮下注射吗啡后痛阈值显著升高,随着给药次数增加,大鼠痛阈值逐渐降低,在第9天降至给药前水平,说明慢性吗啡耐受大鼠模型建立有效;并且,联合应用芬太尼6 μg/kg、12 μg/kg后,可使慢性吗啡耐受大鼠痛阈值增加,部分减轻大鼠吗啡耐受。
δ受体是吗啡作用的靶受体之一,在慢性吗啡耐受形成过程中起着重要作用,主要参于阿片类药物脊髓水平以上的镇痛作用及某些情绪反应,同时也参与μ受体介导的镇痛效应[4]。δ受体在蓝斑核部位分布更为密集。大鼠产生慢性吗啡依赖后,其蓝斑核的超微结构出现变化,主要表现为细胞核膜内陷,粗面内质网颗粒脱落及碎裂,突触前小泡聚积等[5];并且吗啡能够抑制蓝斑核神经元的放电频率,在给予阿片受体阻断剂后,神经元的放电频率得以恢复[6],说明蓝斑核是参与慢性吗啡耐受形成机制中的重要部位。
δ受体是G蛋白偶联受体(GPCR)家族成员,与激动剂结合后,发生磷酸化,与G蛋白结合,G蛋白随之发生构象改变,Gα亚基上的GDP被GTP所取代,δ受体与G蛋白脱离,与胞浆内的β-arrestin 1结合进入细胞内,这一过程为受体脱敏与内吞[7]。受体进入细胞内后,可再通过脱磷酸复敏,回到细胞膜表面,重新恢复和配体结合能力。
本研究结果显示,大鼠产生慢性吗啡耐受后,其蓝斑核δ受体与β-arrestin 1的mRNA与蛋白表达均明显降低,这与Gomes等[1]和Fan等[8]的研究结论是一致的。在吗啡耐受研究方面,有学者认为不同的阿片类药物诱导受体发生脱敏与内吞的水平不同,Whistler等[9]与Alvarez等[10]提出“RA/VE值”概念,即阿片类药物效能与其诱导受体内吞能力的比值。吗啡的RA/VE值为8.5[2],高于其他阿片类药物如芬太尼、美沙酮等;细胞实验结果证实吗啡诱导依赖GRKs催化受体磷酸化的能力很弱[11],阻碍受体内吞过程,甚至神经元在接受吗啡预处理后,埃托啡诱导的阿片受体内吞受到阻碍[12]。在受体发生内吞过程中,细胞信号蛋白β-arrestin与受体相结合,并使之与G蛋白解偶联是一关键步骤。β-arrestin 1以激动剂增强的方式与δ受体直接作用,而去除δ受体C末端后这种作用被减弱[13]。双链RNAi技术实验证实,β-arrestin 1参与磷酸化δ受体内吞过程[14]。既往研究结果表明,β-arrestin与磷酸化的阿片受体之间具有更高的亲和力,易于与之结合[15]。因此,可以推断由于吗啡的高RA/VE值,诱导依赖GRKs催化受体磷酸化能力很弱,使受体水平磷酸化基团减少,不易与β-arrestin结合,减少可磷酸化的阿片受体数量,并且使β-arrestin 1表达也处于低水平状态。
DAMGO是μ受体激动剂,RA/VE值为1.0,将其给予慢性吗啡耐受大鼠,可减轻大鼠慢性吗啡耐受程度,增强吗啡镇痛效应,说明联合应用低RA/VE值的μ受体激动剂可减缓慢性吗啡耐受的发生[16]。芬太尼是临床常用的阿片类镇痛药,与吗啡相比,不易诱发阿片耐受,它同时作用于μ、δ受体,RA/VE值仅为1.8,与DAMGO相近;在激动μ受体发挥镇痛作用的同时,不影响μ受体内吞进入细胞[17],但对δ受体是否具有相同作用尚无报道。本实验中,为避免联合使用阿片类药物可能产生的副作用,芬太尼使用剂量为3 μg/kg、6 μg/kg、12 μg/kg,均低于大鼠芬太尼 ED5013 μg/kg。实验结果表明,芬太尼 6 μg/kg、12 μg/kg使慢性吗啡耐受大鼠痛阈值增加,说明大鼠慢性吗啡耐受程度有所减轻;但芬太尼6 μg/kg、12 μg/kg并没有上调蓝斑部位δ受体mRNA及蛋白表达,也就是说δ受体数量并无增加,说明小剂量芬太尼无法通过上调δ受体表达延缓慢性吗啡耐受产生,δ受体脱敏与内吞过程可能受阻;但在芬太尼6 μg/kg、12 μg/kg作用下,大鼠蓝斑核β-arrestin 1 mRNA及蛋白表达有所上调,而相应地δ受体表达却没有增加,推测主要可能有以下两种原因:一是由于吗啡的高RA/VE值,减少δ受体磷酸化基团,使可磷酸化δ受体数量降低;二是与δ受体的代谢途径有关,该受体内吞进入细胞内,大部分快速进入溶酶体被蛋白酶降解[18]。芬太尼3 μg/kg无上述作用,可能与使用剂量过低、无法有效激动受体有关。
本实验中仍存在许多有待深入研究的地方。在阿片受体的脱敏与内吞过程中除β-arrestin 1之外,GRKs也发挥重要作用,值得我们关注。此外,细胞表面的阿片受体的磷酸化位点、受体之间的相互作用也需进一步探讨。
综上所述,芬太尼6 μg/kg、12μ g/kg可增强吗啡镇痛作用,部分延缓大鼠慢性吗啡耐受产生,其蓝斑部位β-arrestin 1 mRNA及蛋白表达水平上调参与这一过程,但详尽作用机制尚需进一步研究。
[1]Gomes I,Gupta A,Filipovska J,et al.A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia[J].Proc Natl Acad Sci USA,2004,101(9):5135-5139.
[2]Hashimoto T,Saito Y,Yamada K,et al.Enhancement of morphine analgesic effect with induction of mu-opioid receptor endocytosis in rats[J].Anesthesiology,2006,105(3):574-580.
[3]Emmett-Oglesby MW,Shippenberg TS,Herz A,et al.Fentanyl and morphine discrimination in rats continuously infused with fentanyl[J].Behav Pharmacol,1989,1(1):3-11.
[4]Simonin F,Befort K,Gavériaux-Ruff C,et al.The human delta-opioid receptor:genomic organization,cDNA cloning,functional expression,and distribution in human brain[J].Mol Pharmacol,1994,46(3):1015-1021.
[5]Miao H,Qin BY,Yang Y,et al.Ultrastructural changes in rat locus coeruleus by chronic opioids[J].Acta Neuropathol Berl,1997,94(2):109-115.
[6]Holland LN,Shuster JJ,Buccafusco,et al.Role of spinal and superspinal muscarinic receptor in the expression of morphine withdrawal symptoms in the rat[J].Neuropharmacology,1993,32(12):1387-1395.
[7]Müller W,Hallermann S,Swandulla D,et al.Opioidergic modulation of voltage-activated K+currents in magnocellular neurons of the supraoptic nucleus in rat[J].J Neurol Physiol,1999,81(7):1617-1625.
[8]Fan XL.Differential regulation of β-arrestin 1 and β-arrestin 2 gene expression in rat brain by morphine[J].Neuroscience,2003,117(11):383-389.
[9]Whistler JL,Enquist J,Marley A,et al.Modulation of postendocytic sorting of G protein-coupled receptors[J].Science,2002,297(21):615-620.
[10]Alvarez V,Arttamangkul S,Williams JT.A RAVE about opioid withdrawal[J].Neuron,2001,32(8):761-763.
[11]Zhang J,Ferguson SS,Barak LS,et al.Role for G protein coupled receptor kinase in agonist-specific regulation of mu opioid receptor responsiveness[J].Proc Natl Acad Sci USA,1998,95(12):7157-7162.
[12]Koch T,Schulz S,Pfeiffer M,et al.C-terminal splice variants of the mouse mu-opioid receptor differ in morphine induced internalization and receptor resensitization[J].J Biol Chem,2001,276(10):31408-31414.
[13]Cen B,Xiong Y,Ma L,et al.Direct and differential interaction of beta-arrestins with the intracellular domains of different opioid receptors[J].Mol Pharmacol,2001,59(5):758-764.
[14]Zhang X,Wang F,Chen X,et al.Beta-arrestin 1 and beta-arrestin 2 are differentially required for phosphorylation-dependant and-independent internalization of delta-opioid receptors[J].J Neurochem,2005,95(6):169-178.
[15]Gurevich V,Gurevich EV.The molecular acrobatics of arrestin activation[J].Trends Pharmacol Sci,2004,25(7):105-111.
[16]Whistler JL,He Li,Fong J.Regulation of Opioid Receptor Trafficking and morphine tolerance by receptor oligomerization[J].Cell,2003,108(4):271-282
[17]Zuo Z.The role of opioid receptor internalization and β-arrestins in the development of opioid tolerance[J].Anesth Analg,2005,101(5):728-734.
[18]Morinville A,Cahill CM,Esdaile MJ,et al.Regulation of delta-opioid receptor trafficking via mu-opioid receptor stimulation:evidence from mu-opioid receptor knock-out mice[J].J Neurosci,2003,23(6):4888-4898.
猜你喜欢
当代医药论丛(2022年3期)2023-01-04
介入放射学杂志(2022年10期)2022-11-02
中国中医药信息杂志(2022年6期)2022-06-27
科技创新与应用(2021年7期)2021-02-04
上海医学(2020年2期)2020-12-31
医药前沿(2020年23期)2020-12-03
智富时代(2019年6期)2019-07-24
智富时代(2019年6期)2019-07-24
中国药理学与毒理学杂志(2017年4期)2017-06-01
中国比较医学杂志(2017年10期)2017-01-16
