
附子多糖保护缺氧/复氧乳鼠心肌细胞及其抗内质网应激的机制研究*
2012-11-06 04:03吴伟康
中国病理生理杂志 2012年3期
刘 颖, 纪 超, 吴伟康
(1广东药学院基础学院病理生理学教研室,广东 广州 510006; 2中山大学中西医结合研究所,广东 广州 510089)
1000-4718(2012)03-0459-05
2011-10-12
2011-12-05
广东省科技计划项目(No.2008B060600055)
△通讯作者Tel:020-39352121; E-mail:13322819916@163.com
附子多糖保护缺氧/复氧乳鼠心肌细胞及其抗内质网应激的机制研究*
刘 颖1△, 纪 超1, 吴伟康2
(1广东药学院基础学院病理生理学教研室,广东 广州 510006;2中山大学中西医结合研究所,广东 广州 510089)
目的观察附子多糖对缺氧/复氧后心肌细胞的保护,并探讨附子多糖的保护机制是否与其抑制内质网应激反应有关。方法建立乳鼠心肌细胞缺氧/复氧模型,将乳鼠心肌细胞分为正常对照组、缺氧/复氧组(缺氧3 h后复氧6 h)和不同浓度附子多糖(0.1 g/L、1 g/L、10 g/L、20 g/L)+缺氧/复氧组。MTT法测定心肌细胞存活率,流式细胞术检测心肌细胞凋亡率,Western blotting分析葡萄糖调节蛋白78(GRP78)、CCAAT增强子结合蛋白同源蛋白(CHOP)及caspase-12的表达,荧光定量PCR进一步检测CHOP及caspase-12 mRNA表达。结果缺氧复氧后,心肌细胞中内质网应激反应标志蛋白GRP78表达增加,内质网应激凋亡相关蛋白CHOP及caspase-12蛋白和mRNA表达亦明显升高。与缺氧/复氧组相比较,附子多糖预处理24 h后可以有效抑制缺氧/复氧引起的GRP78、 CHOP和caspase-12的表达上调,增加心肌细胞的存活率,抑制心肌细胞凋亡的发生。附子多糖的保护效应呈剂量依赖形式,10 g/L剂量时达到峰值。结论附子多糖保护缺氧/复氧后心肌细胞的可能机制与其抑制内质网应激所介导的细胞凋亡途径相关。
附子多糖; 内质网应激; 缺氧/复氧损伤; 心肌细胞
心肌缺血再灌注损伤是临床上常见的病理过程,其发生机制复杂,涉及多方面因素[1]。近年来研究证实,心肌缺血再灌注过程中引发的氧化应激、钙超载、ATP耗竭等将诱发内质网应激(endoplasmic reticulum stress,ERS),过度ERS介导的心肌细胞功能的障碍及心肌细胞凋亡进一步参与了缺血再灌注损伤的发生[2]。附子多糖(Fuzi polysaccharide,FPS)是从中药附子中提取分离的多糖成分,新近研究提示,附子多糖具有良好的抗心肌氧化应激损伤的作用[3]。但附子多糖能否对抗内质网应激发挥对心肌的保护,尚未见报道。由于缺氧和缺血有着极其相似的病理改变和发病机制,本实验以缺氧/复氧乳鼠心肌细胞模型模拟在体心肌缺血再灌注损伤,观察FPS对缺氧/复氧心肌细胞的保护,和对内质网应激标志蛋白78(glucose-regulated protein 78,GRP78)、内质网相关促凋亡蛋白 (CCAAT/enhancer-binding protein homologous protein,CHOP)和cas-pase-12表达的影响,探讨附子多糖能否通过抑制ERS来保护心肌细胞,对抗缺氧/复氧引起的损伤,从而为深入阐明附子多糖的药理作用提供新的实验资料。
材 料 和 方 法
1材料
1.1药物 鉴定合格的附子多糖由中山大学中西医结合研究所提供[4],分子量14 kD,纯度大于99.8%。使用时溶于细胞培养液中配成实验所需浓度。
1.2动物 出生48 h内的SD乳鼠(购于广州中医药大学实验动物中心,动物合格证号为SCXK 粤2008-0020)。
1.3主要试剂 胰蛋白酶(BioSHARP),Ⅱ型胶原酶(Sigma),青-链霉素溶液(吉诺生物医药技术有限公司);DMEM高糖培养基及DMEM低糖培养基(Hyclone);胎牛血清(杭州四季青生物工程有限公司);AnnexinⅤ-FITC试剂(Beckman Coulter);兔抗大鼠GRP78、CHOP、caspase-12及GAPDH抗体(Cell Signaling Technology);SDS-PAGE凝胶配制试剂盒、胞浆蛋白抽提试剂盒(碧云天生物科技研究所);WB超敏发光液(北京普利来基因有限公司);Trizol、M-MLV逆转录酶和RNasin (Invitrogen),Thunderbird SYBR®Green qPCR Mix(QPS-201)(Toyobo)。
1.4仪器 ELx800酶标仪(BioTek);Centrifuge 5804R台式冷冻离心机(Eppendorf);Coulter Epics XL流式细胞仪(Beckman Coulter);MiniOpticon Real-time PCR仪(Bio-Rad);垂直电泳仪及转膜仪(C.B.S. Scientific);凝胶成像系统(Vilber Lourmat);CO2培养箱(Model TC2323)(Shel Lab)。
2方法
2.1乳鼠心肌细胞原代培养 无菌操作取出乳鼠心脏,剪碎,消化法分离心肌细胞。收集消化液后经200目筛过滤制成单细胞悬液,离心弃上清,用含20%胎牛血清的高糖DMEM培养液重悬细胞,接种于培养瓶中,差速贴壁培养90 min,去除成纤维细胞。调节细胞浓度至5×108/L接种于96孔板,置于CO2培养箱培养,48 h后换含15%胎牛血清的高糖DMEM培养液,待细胞基本融合成片,选择生长良好、搏动规律的细胞用于实验。
2.2心肌细胞缺氧/复氧模型的建立 不含血清的DMEM低糖培养基,纯氮1 L/min流量充分饱和30 min,用上述培养基置换正常培养基,将细胞放入密封袋充入高纯度氮气,密封放入培养箱培养为缺氧;用含15%胎牛血清的高糖DMEM培养液置换培养基,置于95%O2+5%CO2培养箱常氧培养为复氧。
2.3实验分组 将心肌细胞分为3组:(1)正常对照(Control)组:用含15%血清的DMEM高糖培养基常规培养9 h;(2)缺氧/复氧(H/R)组:缺氧3 h后复氧6 h;(3)附子多糖+缺氧/复氧(FPS+H/R) 组:将心肌细胞置入不同浓度的(0.1 g/L、1 g/L、10 g/L、20 g/L)附子多糖培养液中,常规培养24 h后,缺氧3 h后复氧6 h。
2.4MTT法检测细胞活力 取对数生长期细胞接种于96孔板内,实验结束后,每孔加入5 mg/L噻唑蓝(methylthiazolyldiphenyl tetrazolium bromide,MTT)溶液20 μL,37 ℃培养4 h,吸去孔内液体,每孔加入二甲基亚砜(dimethyl sulfoxide, DMSO)150 μL,摇床10 min,待结晶充分溶解后,590 nm波长酶标仪检测光吸收值。细胞存活率(%)=处理组A590/正常组A590×100%。
2.5细胞凋亡率 实验结束后,收集细胞,用0.02%EDTA消化细胞,制备细胞悬液,用Annexin V-FITC试剂盒中binding buffer悬浮细胞,调节细胞浓度为1×109cell/L,取100 μL细胞,分别加入碘化丙啶(propidium iodide,PI) 和膜联蛋白V(Annexin V),避光冰浴10 min,上机检测。
2.6GRP78、CHOP和caspase-12的Western blotting分析 收集细胞,提取细胞总蛋白, Bradford 法测蛋白浓度。取样品蛋白质 30 μg行SDS-PAGE凝胶电泳,转移蛋白至硝酸纤维膜,5%封闭液封闭2 h,分别加入GRP78(1∶200)、CHOP(1∶200)和caspase-12(1∶200)抗体,和上样对照GADPH抗体(1∶400),4 ℃孵育过夜。加入Ⅱ抗(1∶1 000),37 ℃振荡孵育1 h。 ECL显色,放射自显影检测。以GAPDH作内参照,分析GRP78、CHOP和 casepase-12蛋白条带扫描灰度变化。
2.7荧光定量PCR测定CHOP和caspase-12 mRNA的表达 提取心肌细胞总RNA。所有实验用品均预先进行去RNA酶处理, 整个过程在冰浴中进行。采用逆转录酶及随机引物进行逆转录反应, 严格按逆转录试剂盒说明书操作。实时荧光定量PCR引物由上海英俊公司合成。CHOP上游引物5’-TGTTGAAGATGAGCGGGTG-3’,下游引物5’-CAAGGTGAAAGGCAGGGACTC-3’;Casepase-12序列,上游引物5’-ATTCCTGGTCTTTATGTCCC-3’,下游引物5’-ATACTCTCTCAATGGTGGGC-3’;β-actin上游引物5’-CTATTGGCAACGAGCGGTT-3’,下游引物5’-GGCATAGAGGTCTTTACGGATGT-3’。采用高温启动法进行实时荧光定量PCR循环, 扩增的每个标准均设内参对照, 每个指标均有正常组作对照。进行PCR 循环扩增(40个循环) , 循环条件(两步法, 退火60 ℃,延伸72 ℃)。取相对定量( relative quantity, RQ)值进行统计分析。
3统计学处理
结 果
1心肌细胞存活率
与正常对照组相比,缺氧/复氧组的细胞存活率显著下降(P<0.01)。经FPS预处理24 h后,与缺氧/复氧组相比, FPS呈剂量依赖性增加心肌细胞的存活率,在10 g/L时基本达到饱和,20 g/L时心肌细胞存活率增幅趋缓,见表1。
表1各组心肌细胞存活率、凋亡率以及CHOPmRNA和casepase-12mRNA表达量的比较


GroupDose(g/L)Survivalrate(%)Apoptoticrate(%)CHOPmRNARQCaspase-12mRNARQControl-1004.80±0.65 11H/R-52.32±2.90**18.09±1.66**1.76±0.29**1.87±0.31**FPS+H/R2072.58±2.31△△14.26±1.98△△1.29±0.09△△1.32±0.25△△FPS+H/R1072.41±2.52△△14.17±1.11△△1.31±0.12△△1.33±0.19△△FPS+H/R168.97±2.33△△16.99±1.66△1.57±0.08△1.62±0.17△FPS+H/R0.158.62±2.14△17.92±1.451.72±0.271.86±0.33
**P<0.01vscontrol group;△P<0.05,△△P<0.01vsH/R group.
2心肌细胞凋亡率
缺氧/复氧后,心肌细胞出现大量凋亡,与正常对照组相比有显著差异(P<0.01)。经不同浓度的FPS预先处理后,FPS可剂量依赖地减少缺氧/复氧所造成的细胞凋亡,1 g/L时开始具有统计学意义,10 g/L时达到峰值,见表1、图1。
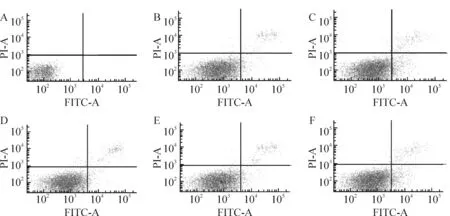
Figure 1. Flow cytometry showed the changes of cell apoptosis among various groups.A:control; B:H/R; C~F:FPS (20, 10, 1 and 0.1 g/L, respectively)+H/R.
图1流式细胞术检测各组心肌细胞凋亡的变化
3GRP78、CHOP和caspase-12的蛋白表达
Western blotting结果显示,与正常对照组相比,缺氧/复氧后GRP78、CHOP和caspase-12蛋白表达均明显增加。不同浓度的FPS预处理,1 g/L浓度以上时可以抑制GRP78、CHOP和caspase-12的表达,与缺氧复氧组对比,差异有统计学意义。在10 g/L时,对CHOP和caspase-12蛋白表达的抑制基本饱和,见图2。
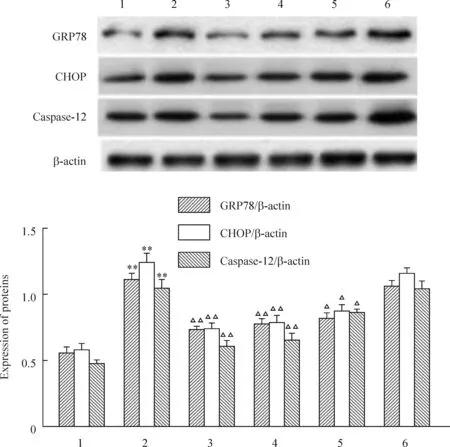

图2各组心肌细胞GRP78、CHOP和casepase-12蛋白表达的比较
4CHOPmRNA和caspase-12mRNA表达
缺氧/复氧后CHOP mRNA及caspase-12 mRNA表达明显增加(P<0.01)。FPS干预后,FPS组与缺氧/复氧组相比,可剂量依赖地抑制CHOP mRNA及caspase-12 mRNA表达,浓度达到1 g/L时差异具有统计学意义,10 g/L时达到峰值,见表1。
讨 论
内质网是细胞内蛋白质合成折叠、钙储存的重要场所。当在细胞内外因素的影响下,内质网稳态遭到破坏,导致大量错误或者未折叠蛋白质在内质网腔中聚集,激活相应的信号通路,引发细胞内一系列的反应,称为内质网应激[5]。现已发现的内质网信号转导通路包括:PERK (protein kinase R-like ER kinase)通路、ATF6(activating transcription factor 6)通路和IRE1( inositol requiring enzyme 1)通路,转导通路的激活与内质网分子伴侣GRP78结合了错误折叠及未折叠蛋白质后,与3种感应蛋白分离密切相关。因此,GRP78被视为内质网稳态的感受器,其表达上调标志ERS的发生。ERS是细胞的一种自我保护性机制,当严重的或长时间的内质网应激损伤了内质网的功能时,将诱导凋亡通路的激活而触发细胞死亡[6]。ERS是继死亡受体活化、线粒体损伤途径后发现的一条重要的细胞凋亡途径,CHOP基因的激活转录和内质网特有的caspase-12 的激活通路是内质网应激细胞凋亡通路的重要组成[7]。
新近的研究表明,内质网应激及其诱导的凋亡与心肌缺血再灌注损伤的发生发展密切相关。Xin等[6]发现,心肌缺血再灌注能够激活GRP78和CHOP的表达。Zhang等[7]也在离体心脏模型上进一步证实了,缺血再灌注导致 ERS,促进 CHOP 的表达升高和caspase-12 裂解。而CHOP基因缺失小鼠,其再灌注后心肌梗死面积缩小伴随细胞凋亡率的降低[8]。本实验观察到,心肌缺氧复氧后,心肌细胞存活率下降,凋亡率升高,伴随GRP78、CHOP、caspase-12的表达明显增加。分析原因为,缺氧复氧诱导心肌细胞产生ERS,强烈的ERS激活了CHOP、caspase-12,进而引起随后的凋亡信号转导级联反应,导致细胞死亡。
目前,基于内质网应激及其诱导凋亡的分子机制,研发心肌缺血再灌注损伤保护的新策略成为研究的增长点。近年来,中药多糖因为高效低毒的特点成为药物开发的热点。资料表明,许多中药多糖具有提高抗氧化酶活性、清除自由基、抑制脂质过氧化,从而保护生物膜的作用[9]。附子多糖是从附子中提出的一种多糖化合物,具有一个半缩醛羟基,能和活性氧发生反应,直接清除自由基,对处于氧化应激状态的细胞具有保护作用,但目前对其功能的研究报道甚少。刘古锋等[3]证明,予附子多糖处理后,能够明显提高心肌组织抗氧化酶的活性,减少小鼠力竭运动所致的氧化应激损伤,提高小鼠的运动耐力。附子多糖还可以通过上调抗凋亡基因bcl-2的表达,抑制凋亡蛋白酶caspase-3的活化,实现抗凋亡的作用[10]。本实验从细胞水平,观察附子多糖对缺氧复氧乳鼠心肌损伤的影响。结果显示,予以附子多糖预先处理后,附子多糖可以逆转缺氧复氧所造成的心肌细胞存活率的下降和凋亡率的升高,同时,附子多糖抑制GRP78、CHOP、caspase-12蛋白及CHOP、caspase-12 mRNA的表达,附子多糖的保护效应呈剂量依赖性。实验结果说明,附子多糖可以保护心肌细胞对抗缺氧复氧损伤,其作用机制与附子多糖抑制内质网应激反应,维持内质网稳态,阻碍内质网应激诱导的细胞凋亡有关。但附子多糖通过哪些通路作用于内质网,减轻内质网应激,还有待于进一步的研究。
[1] 殷 然,王梦洪,郑泽琪,等.肝X受体抑制乳鼠心肌细胞缺氧/复氧损伤[J].中国病理生理杂志,2011,27(9):1671-1675.
[2] Marciniak S,Ron D. Endoplasmic reticulum stress signaling in disease[J]. Physiol Rev,2006,86(4):1133-1149.
[3] 刘古峰,吴伟康,段新芬. 附子多糖对力竭运动小鼠自由基代谢的影响[J].陕西医学杂志,2008,37(5):529-532.
[4] Zhao C,Li M ,Luo Y, et al. Isolation and structural characterization of an immunostimulating polysaccharide from Fuzi,Aconitumcarmichaeli[J]. Carbohydr Res, 2006, 341(4):485-491.
[5] Rutkowski DT,Kaufman RJ.A trip to the ER: coping with stress [J]. Trends Cell Biol,2004, 14(1):20-28.
[6] Boyce M,Yuan J. Cellular response to endoplasmic reticulum stress:a matter of life or death[J]. Cell Death Differ, 2006, 13(3):363-373.
[7] Glembotski CC. The role of the unfolded protein response in the heart [J]. J Mol Cell Cardiol, 2008, 44(3):453-459.
[8] Qi X, Vallentin A, Churchill E, et al.δPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart[J]. J Mol Cell Cardiol, 2007, 43(4):420-428.
[9] Zhang GG, Teng X, Liu Y, et al. Inhibition of endoplasm reticulum stress by ghrelin protects against ischemia/reperfusion injury in rat heart[J]. Peptides, 2009, 30(6):1109-1116.
[10] Miyazaki Y, Kaikita K, Endo M, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation[J]. Arterioscler Thromb Vasc Biol, 2011, 31(5):1124-1132.
Fuzipolysaccharideprotectsneonatalratcardiomyocyteswithhypoxia-reoxygenationbyinhibitingendoplasmicreticulumstress
LIU Ying1, JI Chao1, WU Wei-kang2
(1DepartmentofPathophysiology,SchoolofBasicCourses,GuangdongPharmaceuticalUniversity,Guangzhou510224,China;2InstituteofIntegratedTraditionalChineseandWesternMedicine,SunYet-senUniversity,Guangzhou510089,China.E-mail: 13322819916@163.com)
AIM: To investigate whether the protection mechanism of Fuzi polysaccharide (FPS) is related to inhibition of endoplasmic reticulum stress in cultured neonatal rat cardiomyocytes with hypoxia/reoxygenation (H/R).METHODSCultured rat myocardial cells were divided into control group, H/R group (hypoxia for 3 h and reoxygenation for 6 h) and different concentrations of FPS (0.1 g/L, 1 g/L, 10 g/L or 20 g/L) +H/R groups. The cell survival was detected by MTT assay and cell apoptosis of cardiomyocytes was measured by flow cytometry using Annexin V-FITC staining. The expression of glucose-regulated protein 78 (GRP78), CCAAT/enhancer-binding protein homologous protein (CHOP) and caspase-12 were determined by Western blotting. The mRNA expression of CHOP and caspase-12 was detected by quantitative PCR.RESULTSAfter reoxygenation, the expression of GRP78, CHOP and caspase-12 in cardiomyocytes was increased. Compared with H/R group, the expression of GRP78, CHOP and caspase-12 in FPS+H/R groups was significantly inhibited, the survival rate of cardiomyocytes was increased and the apoptosis of cardiomyocytes was inhibited. This protective effect of FPS was in a dose-dependent manner and reached its peak at 10 g/L.CONCLUSIONFuzi polysaccharide protects cardiomyocytes from H/R injury. The mechanism is related to inhibiting endoplasmic reticulum stress.
Fuzi polysaccharide; Endoplasmic reticulum stress; Hypoxia/reoxygenation injury; Cardiomyocytes
R541.6
A
10.3969/j.issn.1000-4718.2012.03.013
猜你喜欢
世界科学技术-中医药现代化(2021年7期)2021-11-04
现代临床医学(2021年1期)2021-01-26
山东医药(2021年28期)2021-01-11
Digital Chinese Medicine(2020年3期)2020-12-14
中成药(2018年5期)2018-06-06
中成药(2017年12期)2018-01-19
中成药(2017年12期)2018-01-19
农家科技中旬版(2016年9期)2016-11-02
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国药理学与毒理学杂志(2015年3期)2015-12-16
